ในอุตสาหกรรมยา, คุณภาพและความปลอดภัยของผู้ป่วยเป็นสิ่งสำคัญยิ่ง. ทุกแท็บเล็ต, ขวด, หรือบรรจุภัณฑ์พลาสติกจะต้องผลิตภายใต้การควบคุมที่เข้มงวดเพื่อให้มั่นใจว่ามีประสิทธิภาพ, บริสุทธิ์, และปลอดภัย. นั่นคือสิ่งที่ การปฏิบัติตาม GxP เข้ามา. GxP เป็นคำทั่วไปที่มีความหมายว่า “ดี x ฝึกฝน,” โดยที่ “x” สามารถเป็นการผลิตได้, ห้องปฏิบัติการ, คลินิก, การกระจาย, ฯลฯ. โดยพื้นฐานแล้ว, หลักเกณฑ์ของ GxP ถือเป็นข้อบังคับ ระบบคุณภาพ บริษัทยานั้นๆ จะต้องปฏิบัติตาม เพื่อจัดทำเอกสารและควบคุมกระบวนการทั้งหมด. ด้วยกัน, กฎเหล่านี้ช่วยให้แน่ใจว่ายาได้รับการผลิตและควบคุมให้ได้มาตรฐานคุณภาพสูงอย่างสม่ำเสมอตลอดการพัฒนา, การผลิต, การทดสอบ, บรรจุภัณฑ์, และการกระจายสินค้า.
หน่วยงานกำกับดูแลเช่น อย (เรา. สำนักงานคณะกรรมการอาหารและยา), EMA (สำนักงานยาแห่งยุโรป), WHO, และอื่นๆ บังคับใช้มาตรฐาน GxP. ตัวอย่างเช่น, แนวปฏิบัติที่ดีในการผลิตของ FDA ในปัจจุบัน (ซีจีเอ็มพี) กฎ (21 ชิ้นส่วน CFR 210–211) กำหนดข้อกำหนดขั้นต่ำสำหรับการผลิต, การบรรจุ, และกักเก็บยาให้ปลอดภัยมีส่วนผสมและความแข็งแรงที่ถูกต้อง. ในทำนองเดียวกัน, แนวทางของ WHO และ EU ครอบคลุม GMP/GLP/GDP/GVP ทั่วโลก. ในทางปฏิบัติ, บริษัทยาพัฒนาขึ้น ระบบการจัดการคุณภาพ และขั้นตอนการปฏิบัติงานมาตรฐาน (จิบ) เกี่ยวกับหลักการของ GxP. ผลิตภัณฑ์ทุกชุดมีบันทึกที่สามารถตรวจสอบย้อนกลับได้ตั้งแต่ต้นจนจบ. แม้แต่อุปกรณ์ก็ต้องมีคุณสมบัติ (พร้อมคุณสมบัติการติดตั้ง/การปฏิบัติงาน/ประสิทธิภาพ – IQ/OQ/PQ) และได้รับการตรวจสอบเพื่อให้ตรงตามข้อกำหนดของ GxP.

GxP หมายถึงอะไรในเภสัชภัณฑ์?
คำย่อ GxP ย่อมาจาก แนวปฏิบัติ "x" ที่ดี, ที่ไหน “จี” คือ “ดี” และ “พ” คือ “การฝึกฝน” “x” แสดงถึงสาขาวิชาต่างๆ. GxP ไม่ใช่กฎระเบียบเดียว แต่เป็นกลุ่มแนวทางด้านคุณภาพ. สาขาวิชาหลักของ GxP ได้แก่:
- แนวปฏิบัติที่ดีในการผลิต (GMP): ตรวจสอบให้แน่ใจว่าผลิตภัณฑ์ได้รับการผลิตและควบคุมอย่างสม่ำเสมอตามมาตรฐานคุณภาพ. GMP ครอบคลุมถึงการผลิต, อุปกรณ์, สิ่งอำนวยความสะดวก, บุคลากร, และเอกสารประกอบเพื่อลดการปนเปื้อน, การผสมผสาน, และข้อผิดพลาด.
- แนวปฏิบัติที่ดีในห้องปฏิบัติการ (GLP): ควบคุมการดำเนินการศึกษาในห้องปฏิบัติการที่ไม่ใช่ทางคลินิก (เช่น การทดสอบทางพิษวิทยา) เพื่อให้มั่นใจในความสมบูรณ์ของข้อมูลและความสามารถในการทำซ้ำ. GLP ช่วยให้มั่นใจได้ว่าการทดสอบในห้องปฏิบัติการได้รับการบันทึกไว้อย่างดี, ตรวจสอบได้, และเชื่อถือได้.
- การปฏิบัติทางคลินิกที่ดี (จีซีพี): มาตรฐานการออกแบบระดับสากล, การดำเนินการ, การตรวจสอบ, และรายงานการทดลองทางคลินิก. GCP ปกป้องสิทธิของผู้ป่วย, ความสมบูรณ์ของข้อมูล, และรับรองว่าผลการทดลองมีความน่าเชื่อถือ.
- แนวทางปฏิบัติในการกระจายสินค้าที่ดี (จีดีพี): กำหนดมาตรฐานการจัดเก็บ, การจัดการ, และการขนส่งผลิตภัณฑ์ยา. GDP ช่วยให้มั่นใจได้ว่าคุณภาพยาจะคงอยู่ตลอดห่วงโซ่อุปทาน เป็นต้น, โดยการควบคุมอุณหภูมิ, หลีกเลี่ยงการปนเปื้อน, และการติดตามแบทช์.
- แนวปฏิบัติที่ดีในการเฝ้าระวังด้านเภสัชกรรม (จีวีพี): จัดทำแนวทางสำหรับการติดตามความปลอดภัยของยาและรายงานเหตุการณ์ไม่พึงประสงค์หลังจากวางตลาดผลิตภัณฑ์. GVP ช่วยให้บริษัทยาและหน่วยงานกำกับดูแลตรวจจับได้, ประเมิน, และป้องกันผลข้างเคียงด้านลบ.
แต่ละพื้นที่ GxP ได้รับการบังคับใช้โดยกฎระเบียบหรือแนวปฏิบัติเฉพาะ (เช่น, อย 21 CFR สำหรับ GMP/GLP/GCP, EU EudraLex สำหรับ GMP/GDP/GVP, แนวทางครับ, WHO, ฯลฯ). ในขณะที่รายละเอียดต่างกันออกไป, เป้าหมายร่วมกันคือ เพื่อให้ผลิตภัณฑ์ยามีความปลอดภัย, มีประสิทธิภาพ, และมีคุณภาพสูงในทุกขั้นตอน.
โต๊ะ: สาขาวิชาหลักของ GxP(ประเภทของข้อบังคับ GxP)
| ประเภท GxP |
ชื่อเต็ม |
ระยะวงจรชีวิตผลิตภัณฑ์ |
โฟกัส/วัตถุประสงค์ |
| GLP |
แนวปฏิบัติที่ดีในห้องปฏิบัติการ |
การวิจัยยา |
ควบคุมการศึกษาในห้องปฏิบัติการที่ไม่ใช่ทางคลินิก (เช่น การทดสอบความเป็นพิษ) เพื่อให้มั่นใจในคุณภาพของข้อมูลและความสามารถในการตรวจสอบย้อนกลับ. GLP กำหนดมาตรฐานสำหรับขั้นตอนการปฏิบัติงานในห้องปฏิบัติการ, การบันทึกข้อมูล, และการรายงาน. |
| จีซีพี |
แนวปฏิบัติที่ดีในห้องปฏิบัติการ |
การทดลองทางคลินิก |
มาตรฐานสากลสำหรับการทดลองทางคลินิก (ไอ-จีซีพี). ครอบคลุมการออกแบบการทดลองใช้, ความยินยอมที่ได้รับการบอกกล่าว, การตรวจสอบ, และการรายงานผลที่แม่นยำ. |
| GMP |
การปฏิบัติทางคลินิกที่ดี |
การผลิต |
ตรวจสอบให้แน่ใจว่ายาได้รับการผลิตและควบคุมอย่างสม่ำเสมอในการผลิต/บรรจุภัณฑ์. รวมถึงมาตรฐานสิ่งอำนวยความสะดวก/อุปกรณ์, การฝึกอบรมบุคลากร, การทดสอบคุณภาพ. |
| จีดีพี |
แนวทางปฏิบัติในการกระจายสินค้าที่ดี |
คลังสินค้า & โลจิสติกส์ |
รับประกันการจัดเก็บและการขนส่งยาที่เหมาะสม (เช่น, อุณหภูมิที่ถูกต้อง, การจัดการที่ปลอดภัย) ดังนั้นคุณภาพจึงถูกรักษาไว้ผ่านห่วงโซ่อุปทาน. |
| จีวีพี |
แนวปฏิบัติที่ดีในการเฝ้าระวังด้านเภสัชกรรม |
การติดตามหลังการขาย |
แนวทางการติดตามความปลอดภัยของยาที่วางตลาดอย่างต่อเนื่อง (การรายงานเหตุการณ์ไม่พึงประสงค์, การจัดการความเสี่ยง, การสื่อสารกับเจ้าหน้าที่). |
เหตุใด GxP จึงมีความสำคัญในอุตสาหกรรมยา?
การปฏิบัติตาม GxP ถือเป็นสิ่งสำคัญเพราะว่า ชีวิตขึ้นอยู่กับมันอย่างแท้จริง. วัตถุประสงค์หลักคือ:
- ความปลอดภัยของผู้ป่วย: การปฏิบัติตาม GxP ช่วยลดความเสี่ยง เช่น การปนเปื้อนให้เหลือน้อยที่สุด, การผสมผสาน, หรือข้อผิดพลาดในการใช้ยา. ตัวอย่างเช่น, กฎ GMP (เช่น สุขอนามัยที่เหมาะสมและการควบคุมกระบวนการ) ช่วยป้องกันการปนเปื้อนข้ามระหว่างผลิตภัณฑ์. ไม่มี GxP, ยาที่ไม่ได้มาตรฐานอาจเป็นอันตรายต่อผู้ป่วยหรือขาดประสิทธิภาพได้. หน่วยงานกำกับดูแลเน้นย้ำว่า “กฎ GxP มีอยู่ด้วยเหตุผลเดียว: เพื่อปกป้องผู้ป่วย”.
- คุณภาพและความสม่ำเสมอของผลิตภัณฑ์: กรอบงาน GxP ช่วยให้มั่นใจได้ว่าชุดยาทุกชุดมีคุณสมบัติตรงตามข้อกำหนดเฉพาะเดียวกัน. รวมถึงความแรงที่สม่ำเสมอ, ความบริสุทธิ์, และความมั่นคง. โดยบังคับใช้การควบคุมอย่างเข้มงวด (E.G. การสอบเทียบเครื่องมือ, วิธีการที่ได้รับการตรวจสอบแล้ว, การทดสอบในกระบวนการ), ผู้ผลิตสามารถรับประกันได้ว่าขวดหรือแท็บเล็ตแต่ละขวดตรงกับการอ้างสิทธิ์บนฉลาก.
- การปฏิบัติตามกฎระเบียบและความน่าเชื่อถือ: การปฏิบัติตาม GxP ถือเป็นข้อบังคับตามกฎหมาย. หน่วยงานเช่นอย, EMA, มฮรา, พีเอ็มดีเอ, และ WHO พึ่งพาการตรวจสอบและการตรวจสอบของ GxP. บริษัทที่แสดงให้เห็นถึงแนวปฏิบัติ GxP ที่แข็งแกร่งจะสร้างความเชื่อมั่นกับหน่วยงานกำกับดูแล. การไม่ปฏิบัติตามอาจนำไปสู่จดหมายเตือนได้, ค่าปรับ, การเรียกคืนผลิตภัณฑ์, หรือแย่กว่านั้น. ตามบันทึกคู่มืออุตสาหกรรมฉบับหนึ่ง, หน่วยงานกำกับดูแลดำเนินการตรวจสอบเพื่อ "ยืนยันการปฏิบัติตามมาตรฐาน GxP" ซึ่งเน้นย้ำถึงความสมบูรณ์ของผลิตภัณฑ์และความปลอดภัยของผู้ป่วย.
- ความสมบูรณ์ถูกต้องของข้อมูล: สิ่งสำคัญของ GxP คือการทำให้มั่นใจว่าข้อมูลมีความถูกต้องและเชื่อถือได้. กฎระเบียบที่ทันสมัย (E.G. อย 21 ส่วนซีเอฟอาร์ 11, ภาคผนวกของสหภาพยุโรป 11) กำหนดให้บันทึกทางอิเล็กทรอนิกส์มีความปลอดภัย, ประทับเวลา, และป้องกันการงัดแงะ. หลักการสำคัญเช่น แอลกอฮอล์+ (ระบุแหล่งที่มาได้, ชัดเจน, ร่วมสมัย, ต้นฉบับ, แม่นยำ, บวกเสร็จสมบูรณ์, สม่ำเสมอ, ยั่งยืน, มีอยู่) ใช้เพื่อตัดสินคุณภาพของข้อมูล. ในทางปฏิบัติ, นี่หมายถึงการนำแนวทางการตรวจสอบไปใช้ในซอฟต์แวร์, ล็อคเอกสารหลัก, และตรวจสอบบันทึกชุดงานอย่างสม่ำเสมอ. ความสมบูรณ์ของข้อมูลที่แข็งแกร่งคือรากฐานสำคัญของ GxP; โดยไม่มีมัน, แม้แต่สินค้าที่ทำมาอย่างดีก็ยังไม่เชื่อถือ.
ในระยะสั้น, GxP เป็นรากฐานของการจัดการคุณภาพยา. มันรับประกันได้ว่า ทุกขั้นตอน — ตั้งแต่การทดสอบในห้องปฏิบัติการไปจนถึงการบรรจุขั้นสุดท้าย — ดำเนินการภายใต้การควบคุม, เงื่อนไขที่บันทึกไว้. ตัวอย่างเช่น, ไกด์คนหนึ่งสังเกตเห็นสิ่งนั้น “การปฏิบัติตาม GxP ช่วยให้มั่นใจได้ว่ายาและชีววิทยาได้รับการพัฒนา, ผลิต, และผ่านการทดสอบตามมาตรฐานอันเข้มงวด,- ป้องกันการปนเปื้อนหรือข้อผิดพลาดในการติดฉลากที่มีราคาแพง. ดังนั้น, GxP ไม่เพียงแต่ปกป้องผู้ป่วยเท่านั้น แต่ยังสนับสนุนชื่อเสียงของแบรนด์ที่เชื่อถือได้และการเข้าถึงตลาดสำหรับบริษัทยาอีกด้วย.

ทำความเข้าใจ GMP – มาตรฐาน GxP ที่สำคัญที่สุด
ในขณะที่พื้นที่ GxP ทั้งหมดมีความสำคัญ, GMP (แนวปฏิบัติที่ดีในการผลิต) มักถูกมองว่าเป็นรากฐานสำหรับการปฏิบัติตามข้อกำหนดด้านการผลิตยา. GMP ครอบคลุมกระบวนการผลิตผลิตภัณฑ์ยาทั้งหมด, รวมถึงวัตถุดิบ, อุปกรณ์, สิ่งอำนวยความสะดวก, กระบวนการ, และสายการบรรจุภัณฑ์. เป้าหมายหลักคือการ ลดความเสี่ยง ของการปนเปื้อนหรือการเบี่ยงเบนของผลิตภัณฑ์โดยการบังคับใช้การควบคุมและเอกสารที่เข้มงวด. องค์ประกอบสำคัญของ GMP ได้แก่:
- การออกแบบสิ่งอำนวยความสะดวกและอุปกรณ์: อุปกรณ์ควรได้รับการออกแบบเพื่อการทำงานที่ถูกสุขลักษณะ. ซึ่งหมายถึงการใช้วัสดุเกรดยา (E.G. 316แอล สแตนเลส), รอยเชื่อมเรียบ, ไม่มีโซนตาย, และเข้าถึงทำความสะอาดได้ง่าย. สำหรับเครื่องบรรจุภัณฑ์, สิ่งนี้อาจนำมาซึ่งสายพานลำเลียงแบบคานยื่น, ชิ้นส่วนที่ปล่อยออกมาอย่างรวดเร็ว, และยามที่ปิดล้อม. รูปแบบที่เหมาะสมป้องกันการปะปนกันและอำนวยความสะดวกในการตรวจสอบการทำความสะอาด (ขั้นตอนการทำความสะอาดที่ผ่านการตรวจสอบแล้ว).
- ยูทิลิตี้และการสอบเทียบ: สาธารณูปโภคทั้งหมด (น้ำ, อากาศอัด, ไฟฟ้า) ต้องเป็นไปตามข้อกำหนดด้านคุณภาพ. เครื่องมือและเซ็นเซอร์ (ตาชั่ง, เมตรการไหล, หัววัดอุณหภูมิ) ต้องสม่ำเสมอ การสอบเทียบ ภายใต้ขั้นตอนที่เป็นลายลักษณ์อักษร. FDA คาดหวังอย่างชัดเจนถึงบันทึกการสอบเทียบสำหรับอุปกรณ์ที่สำคัญ. ตัวอย่างเช่น, สายบรรจุขวดจะต้องมีการสอบเทียบปั๊มปริมาตรเพื่อให้แน่ใจว่าแต่ละโดสมีความแม่นยำ.
- การตรวจสอบและการรับรอง: GMP กำหนดให้อุปกรณ์และกระบวนการมีคุณสมบัติและตรวจสอบได้. สิ่งนี้เกี่ยวข้องกับคุณสมบัติการติดตั้ง (ไอคิว), คุณสมบัติการปฏิบัติงาน (โอคิว), และคุณสมบัติการปฏิบัติงาน (PQ) เพื่อพิสูจน์ว่าระบบทำงานได้ตามที่ตั้งใจไว้ (กล่าวถึงในรายละเอียดด้านล่าง). เช่น, เครื่องบรรจุตุ่มใหม่จะต้องผ่านการทดสอบ IQ/OQ/PQ เพื่อยืนยันอุณหภูมิการปิดผนึก, ความสมบูรณ์ของซีล, และระบบการจัดทำดัชนีตรงตามข้อกำหนด. ทุกเครื่องจักรที่สำคัญ (ฟิลเลอร์แคปซูล, กล่องกระดาษ, สายการบรรจุของเหลว) ต้องเป็นไปตามวงจรการตรวจสอบนี้.
- ขั้นตอนการปฏิบัติงานมาตรฐาน (จิบ): รายละเอียด, SOP ที่เป็นลายลักษณ์อักษรมีผลบังคับใช้สำหรับขั้นตอนการผลิต, ทำความสะอาด, การซ่อมบำรุง, และการควบคุมคุณภาพ. ผู้ปฏิบัติงานต้องได้รับการฝึกอบรมเกี่ยวกับ SOP เหล่านี้ และการเปลี่ยนแปลงใดๆ จะต้องได้รับการควบคุมผ่านระบบควบคุมการเปลี่ยนแปลงอย่างเป็นทางการ. ผู้ตรวจสอบจะมองหา SOP และบันทึกการฝึกอบรมที่เป็นปัจจุบัน.
- แนวปฏิบัติที่ดีด้านเอกสาร: GMP เน้นย้ำว่า “ถ้าไม่มีเอกสาร., มันไม่ได้เกิดขึ้น” ทุกขั้นตอน, บันทึกแบทช์, บันทึกการทำความสะอาด, และการทดสอบ QC จะต้องบันทึกให้ชัดเจนและพร้อมกัน. บันทึกการผลิตเป็นชุดที่เสร็จสมบูรณ์ (กทม) จะต้องรวมถึงการกระทบยอดวัสดุ, การตั้งค่าอุปกรณ์, การตรวจสอบระหว่างดำเนินการ, และการเบี่ยงเบน/การแก้ไขใดๆ. แนวโน้มสมัยใหม่ผลักดันบันทึกชุดอิเล็กทรอนิกส์ (อีบีอาร์) โดยมีเส้นทางการตรวจสอบอยู่ข้างใต้ 21 ส่วนซีเอฟอาร์ 11.
- การควบคุมคุณภาพ (การควบคุมคุณภาพ) และการเปิดตัวเป็นชุด: การทดสอบคุณภาพ (E.G. ความแรง, ความเป็นหมัน, บัตรประจำตัว) ดำเนินการกับวัตถุดิบและผลิตภัณฑ์สำเร็จรูป. หลังจากที่แผนก QA/QC ตรวจสอบเอกสารทั้งหมดและผลการทดสอบเท่านั้นจึงจะเผยแพร่เป็นชุด. การตรวจสอบขั้นสุดท้ายนี้ถือเป็นส่วนสำคัญของ GMP.
บล็อกของ Jinlu Packing เกี่ยวกับบรรจุภัณฑ์ GMP เน้นย้ำข้อกำหนดหลายประการเหล่านี้. ตัวอย่างเช่น, มันตั้งข้อสังเกตว่า การบำรุงรักษาเชิงป้องกันและการสอบเทียบ ได้รับคำสั่งจาก GMP (21 ซีเอฟอาร์ 211.68 ต้องมีช่วงเวลาและบันทึกที่กำหนดไว้). นอกจากนี้ยังเป็นการเน้นย้ำ ระบบอัตโนมัติ & การป้องกันข้อผิดพลาด: สายการบรรจุหีบห่อ GMP สมัยใหม่ใช้การตรวจสอบด้วยภาพ, การสแกนบาร์โค้ด, และเชื่อมต่อกันเพื่อหลีกเลี่ยงข้อผิดพลาดของมนุษย์. ความสมบูรณ์ถูกต้องของข้อมูลและการควบคุมคอมพิวเตอร์ยังอยู่ภายใต้ GMP อีกด้วย: ระบบคอมพิวเตอร์ออนไลน์ทั้งหมด (E.G. PLC, ระบบการมองเห็น, HMI) จะต้องปฏิบัติตาม 21 ส่วนซีเอฟอาร์ 11 – หมายถึงการเข้าสู่ระบบที่ไม่ซ้ำใคร, เส้นทางการตรวจสอบ, ลายเซ็นอิเล็กทรอนิกส์, และการเก็บบันทึกที่ปลอดภัย. ในทางปฏิบัติ, ซึ่งหมายความว่าซอฟต์แวร์ควบคุมของเครื่องจะบันทึกทุกการเปลี่ยนแปลงพารามิเตอร์ด้วยการประทับเวลาและ ID ผู้ใช้, และจะต้องมีลายเซ็นอิเล็กทรอนิกส์ของผู้จัดการเพื่ออนุมัติชุดงาน.
รายการตรวจสอบ GMP สำหรับอุปกรณ์บรรจุภัณฑ์
ช่วยในการดูข้อกำหนด GMP ในแบบฟอร์มรายการตรวจสอบ. สำหรับเครื่องจักรบรรจุภัณฑ์, ผู้ผลิตมักจะมั่นใจ:
- ออกแบบ & วัสดุ: การก่อสร้างที่ถูกสุขลักษณะ (ชิ้นส่วนสแตนเลส, ซีลที่ได้รับการรับรองจาก FDA), ไม่มีพื้นที่ "ตาย", พื้นผิวลาดเอียง, ถอดประกอบทำความสะอาดง่าย.
- การตรวจสอบ: ทำ IQ/OQ/PQ ให้สมบูรณ์ในแต่ละเครื่อง. การทดสอบฟังก์ชั่นที่บันทึกไว้ (การปิดผนึก, การกรอก, การชั่งน้ำหนัก) และประสิทธิภาพ. (ดูส่วนคุณสมบัติอุปกรณ์ด้านล่าง)
- การทำความสะอาด: ขั้นตอนการทำความสะอาดที่ผ่านการตรวจสอบแล้วด้วยการทดสอบการเช็ดหรือการล้าง (โดยทั่วไปจะใช้ TOC หรือการตรวจวิเคราะห์เฉพาะ) และบันทึก.
- การซ่อมบำรุง & การสอบเทียบ: ตารางการบำรุงรักษาเชิงป้องกันพร้อมบันทึก. การสอบเทียบปั๊มสูบจ่าย, ตาชั่ง, เซ็นเซอร์เพื่อความแม่นยำ.
- การควบคุม & ระบบอัตโนมัติ: ระบบตรวจสอบด้วยภาพ (เช่น, เครื่องตรวจจับโลหะ, เครื่องตรวจสอบน้ำหนักบนสายพาน) เพื่อตรวจจับข้อบกพร่อง, เซ็นเซอร์เพื่อป้องกันการป้อนผิดพลาด, ลูกโซ่หยุดถ้าประตูเปิด.
- ความสมบูรณ์ถูกต้องของข้อมูล: 21 ส่วนซีเอฟอาร์ 11 การปฏิบัติตาม – เส้นทางการตรวจสอบ, การควบคุมการเข้าถึงของผู้ใช้, ลายเซ็นอิเล็กทรอนิกส์, ข้อมูลที่ปลอดภัย. บันทึกทั้งหมด (จิบ, ข้อมูลแบทช์, การเบี่ยงเบน) เก็บไว้อย่างไม่เปลี่ยนแปลง.
- การตรวจสอบย้อนกลับ: รองรับการทำให้เป็นอนุกรม/UID, บาร์โค้ด, การเชื่อมโยงแต่ละแพ็คหลักเข้ากับบันทึกแบบแบตช์, เปิดใช้งานการเรียกคืนหากจำเป็น. (เครื่อง Jinlu มักจะรวมเครื่องพิมพ์ฉลากหรือเครื่องอ่านรหัสเข้าด้วยกันเพื่อให้สามารถตรวจสอบย้อนกลับได้)
- ด้านสิ่งแวดล้อม & การควบคุมสาย: มาตรฐานห้องสะอาดที่เหมาะสมหากจำเป็น, การตรวจสอบระยะห่างบรรทัดที่จัดทำเป็นเอกสารระหว่างชุดงาน, การติดฉลากที่เหมาะสมและการจัดการวัสดุเพื่อป้องกันการปะปนกัน.
โดยปฏิบัติตามการควบคุมเหล่านี้, ผู้ผลิตสามารถมั่นใจได้ว่าอุปกรณ์บรรจุภัณฑ์ของตนจะทำงานภายใต้หลักเกณฑ์ GMP. ตัวอย่างเช่น, เครื่องเติมแคปซูล และ เครื่องบรรจุพุพอง จาก Jinlu สร้างขึ้นด้วยคุณสมบัติที่พร้อมใช้งาน GMP (พื้นผิว GMP เรียบ, ความสามารถของซีไอพี, ฯลฯ) เพื่อตอบสนองความต้องการเหล่านี้.

ข้อกำหนด GxP สำหรับผู้ผลิตอุปกรณ์ทางเภสัชกรรม
ซัพพลายเออร์อุปกรณ์บรรจุภัณฑ์มีบทบาทสำคัญใน GxP. ผู้ซื้อคาดหวังเครื่องจักรที่ไม่เพียงแต่แข็งแกร่งและมีประสิทธิภาพเท่านั้น, แต่ยังสร้างขึ้นเพื่ออำนวยความสะดวกในการปฏิบัติตามกฎระเบียบอีกด้วย. ข้อกำหนดที่สำคัญได้แก่:
- การสนับสนุนคุณสมบัติ (IR/WH/PQ): ผู้ขายควรจัดให้มี โปรโตคอล และความช่วยเหลือด้านคุณสมบัติ. นี่หมายถึงคุณสมบัติการติดตั้งที่ได้รับการบันทึกไว้ (ไอคิว) เพื่อแสดงว่าเครื่องได้รับการติดตั้งอย่างถูกต้อง, คุณสมบัติการปฏิบัติงาน (โอคิว) เพื่อพิสูจน์ว่ามันใช้งานได้ตามข้อกำหนด, และคุณสมบัติการปฏิบัติงาน (PQ) เพื่อตรวจสอบให้แน่ใจว่าได้ผลลัพธ์ที่ยอมรับได้อย่างสม่ำเสมอ. ตัวอย่างเช่น, Jinlu Packing มอบแต่ละเครื่องด้วยชุด IQ/OQ/PQ และโปรโตคอล FAT/SAT เต็มรูปแบบ. เทมเพลตเหล่านี้มักจะปรับเปลี่ยนได้โดยทีม QA ของผู้ซื้อ, ประหยัดเวลาระหว่างการตรวจสอบ. ผู้จำหน่ายอาจมีส่วนร่วมในการดำเนินการตรวจสอบคุณสมบัติหรือจัดทำรายการตรวจสอบอุปกรณ์ที่ได้รับการรับรอง.
- แพคเกจเอกสาร: พร้อมทั้งตัวเครื่อง, ซัพพลายเออร์ควรจัดส่งเอกสารครบชุด. รายการทั่วไป ได้แก่ คู่มือการใช้งาน, รายการชิ้นส่วนหลัก, แผนงานไฟฟ้า, และ คำแนะนำในการบำรุงรักษา. ที่สำคัญ., อ้วน (การทดสอบการยอมรับจากโรงงาน) และ นั่ง (การทดสอบการยอมรับไซต์) รายงานเอกสารว่าเครื่องผ่านการทดสอบจากโรงงานและนอกสถานที่. ควรมีใบรับรองการสอบเทียบสำหรับอุปกรณ์ตรวจวัดใดๆ ไว้ด้วย. ในทางปฏิบัติ, แพคเกจเอกสารที่เป็นไปตามข้อกำหนดจะแสดงข้อกำหนดของระบบด้วย, SOP การทำความสะอาด, การประเมินความเสี่ยง, และประวัติการควบคุมการเปลี่ยนแปลงใดๆ.
- คุณสมบัติความสมบูรณ์ของข้อมูล: อุปกรณ์สมัยใหม่ควรมีการควบคุมแบบอิเล็กทรอนิกส์ที่สอดคล้องกับมาตรฐานข้อมูล GxP. ซึ่งรวมถึงบัญชีผู้ใช้ที่ปลอดภัยด้วย (การเข้าถึงตามบทบาท), ลายเซ็นอิเล็กทรอนิกส์ที่จำเป็นสำหรับการดำเนินการที่สำคัญ, และเส้นทางการตรวจสอบเต็มรูปแบบของการเปลี่ยนแปลงพารามิเตอร์. ตัวอย่างเช่น, หน้าจอ HMI อาจต้องเข้าสู่ระบบของหัวหน้ากะเพื่อเริ่มการผลิต, และการเปลี่ยนแปลงทุกสูตรหรือการตั้งค่าจะมีการประทับเวลา. เครื่องจักรอาจรองรับเอาท์พุตการบันทึกแบบดิจิทัลด้วย, บูรณาการกับระบบ MES/ERP.
- เครื่องมือตรวจสอบและทดสอบ: ผู้จำหน่ายบางรายมีเครื่องมือซอฟต์แวร์สำหรับการบันทึกข้อมูล, การสอบเทียบ, หรือการตรวจสอบ. นี่อาจเป็นซอฟต์แวร์ที่ติดตั้งไว้ล่วงหน้าสำหรับดำเนินการทดสอบเซ็นเซอร์, หรือฟังก์ชันในตัวเพื่อล็อคพารามิเตอร์ระหว่างการรัน PQ. คุณลักษณะเหล่านี้ช่วยลดความพยายามด้วยตนเองระหว่างการตรวจสอบความถูกต้อง.
- การออกแบบที่ถูกสุขลักษณะและความปลอดภัย: อุปกรณ์จะต้องง่ายต่อการทำความสะอาดและบำรุงรักษา. คุณสมบัติเช่นชิ้นส่วนแบบปลดเร็ว, โซนปลอดสินค้า, และซีไอพี (ทำความสะอาดในสถานที่) ตัวเลือกช่วยให้เป็นไปตามการตรวจสอบการทำความสะอาด. วัสดุที่สัมผัสกับผลิตภัณฑ์ควรเป็นวัสดุเฉื่อย (E.G. 316แอลเอสเอส, พลาสติกที่ได้รับการรับรองจาก FDA). เจ้าหน้าที่รักษาความปลอดภัยและลูกโซ่ช่วยปกป้องผู้ปฏิบัติงาน, แต่ยังรับประกันการปฏิบัติตาม (E.G. เครื่องหยุดทำงานเมื่อเปิด).
- การสนับสนุนหลังการขาย: การปฏิบัติตาม GxP ยังดำเนินอยู่. ผู้ผลิตอาจต้องมีการตรวจสอบคุณสมบัติใหม่หรือการสอบเทียบใหม่เป็นระยะ. ซัพพลายเออร์ควรเสนอบริการตลอดอายุการใช้งาน: อะไหล่ (เพื่อการเปลี่ยนชิ้นส่วนที่ผ่านการตรวจสอบอย่างรวดเร็ว), สัญญาการบำรุงรักษา, และอัปเดตเอกสารการตรวจสอบหากมีการเปลี่ยนแปลงเกิดขึ้น. ความเต็มใจของซัพพลายเออร์ในการให้บริการด้านการรับรอง ณ สถานที่ (IR/WH/PQ) สามารถทำให้ความพยายามในการปฏิบัติตามกฎระเบียบราบรื่นขึ้นอย่างมาก.
รายการตรวจสอบคุณสมบัติอุปกรณ์: ตารางด้านล่างสรุปขั้นตอนและเอกสารทั่วไปสำหรับการตรวจสอบคุณสมบัติเครื่องจักรทางเภสัชกรรมใหม่:
| เฟส |
กิจกรรมสำคัญ |
เอกสารทั่วไป |
| ข้อกำหนดของผู้ใช้ (สสส) |
กำหนดข้อกำหนดที่สำคัญ (E.G. อัตราการส่งออก, ความแม่นยำ) |
ข้อกำหนดความต้องการของผู้ใช้ |
| คุณสมบัติการออกแบบ (ดีคิว) |
ตรวจสอบการออกแบบของผู้จัดจำหน่ายที่ตรงตาม URS |
รายงานการตรวจสอบข้อมูลจำเพาะการออกแบบ |
| การยอมรับจากโรงงาน (อ้วน) |
การทดสอบฟังก์ชั่นหลักๆ จากโรงงาน, มักจะสะท้อนการทดสอบ IQ/OQ |
รายงานไขมัน |
| คุณสมบัติการติดตั้ง (ไอคิว) |
ยืนยันการติดตั้งที่ถูกต้อง: สาธารณูปโภค, การตั้งค่าทางกล, เอกสารประกอบ (ภาพวาด, ใบรับรอง) |
โปรโตคอลไอคิว & รายการตรวจสอบ |
| คุณสมบัติการปฏิบัติงาน (โอคิว) |
ทดสอบฟังก์ชั่นทั้งหมด: ประสิทธิภาพการทำงานที่ว่างเปล่า, การควบคุม, สัญญาณเตือน, คุณสมบัติด้านความปลอดภัย |
โปรโตคอล OQ & ผลลัพธ์ |
| คุณสมบัติการปฏิบัติงาน (PQ) |
ดำเนินการผลิตเต็มรูปแบบด้วยสินค้าจริง: ตรวจสอบคุณภาพผลผลิต, ความสม่ำเสมอ, สภาวะความเครียด |
โปรโตคอล PQ, เรียกใช้บันทึก, ตัวอย่างผลการทดสอบ |
| การเปิดตัวครั้งสุดท้าย |
ตรวจสอบบันทึกคุณสมบัติทั้งหมด; อนุมัติ QA นำเครื่องจักรเข้าผลิต GMP |
รายงานสรุปคุณสมบัติ |
(บันทึก: เครื่องจักรของ Jinlu มาพร้อมกับ เทมเพลต IQ/OQ/PQ และเต็มรูปแบบ การยอมรับจากโรงงาน แพคเกจเอกสาร, ซึ่งลูกค้าสามารถปรับเปลี่ยนได้ตามต้องการ) แนวทางที่มีโครงสร้างนี้ — ตั้งแต่ความต้องการของผู้ใช้ไปจนถึง PQ — ถูกกำหนดโดยกฎระเบียบเช่น ภาคผนวก GMP ของสหภาพยุโรป 15 และแนวปฏิบัติของอย. การข้ามขั้นตอนใดๆ อาจส่งผลให้เกิดช่องว่างในการปฏิบัติตามข้อกำหนด.

ความท้าทายทั่วไปในการปฏิบัติตามข้อกำหนด GxP
แม้จะมีกฎเกณฑ์ที่ชัดเจน, บริษัทต่างๆ มักเผชิญกับอุปสรรคในการปฏิบัติตาม GxP. ความท้าทายที่พบบ่อยได้แก่:
- การตรวจสอบหรือเอกสารที่ไม่สมบูรณ์: ช่องว่างที่ร้ายแรงที่สุดประการหนึ่งคือการใช้อุปกรณ์ที่ไม่มีบันทึก IQ/OQ/PQ ครบถ้วน. “ข้ามการตรวจสอบ,” หรือมี บางส่วน คุณสมบัติ, ถือเป็นก “ช่องว่าง GMP ร้ายแรง”. ในทำนองเดียวกัน, บันทึกชุดงานและ SOP ที่ขาดหายไปหรือเลอะเทอะจะบ่อนทำลายการปฏิบัติตามข้อกำหนด: ผู้ตรวจสอบได้รับการฝึกอบรมให้ค้นหาเอกสาร. ดังที่ผู้เชี่ยวชาญท่านหนึ่งกล่าวไว้: “ถ้ามันไม่ได้เขียนลงไป, มันไม่ได้เกิดขึ้น” การเก็บบันทึกไม่ดี (ไฟล์ที่สูญหาย, บันทึกที่อ่านไม่ออก, รุ่นที่ล้าสมัย) เป็นธงสีแดงทั่วไป.
- การบำรุงรักษาอุปกรณ์และการสอบเทียบสิ้นสุดลง: หน่วยงานกำกับดูแลมักพบปัญหาต่างๆ เช่น วันครบกำหนดการสอบเทียบที่หมดอายุ หรือการเลื่อนการซ่อมแซม. เซ็นเซอร์ที่เสียหายหรือเครื่องชั่งที่ไม่ได้สอบเทียบจะนำไปสู่ข้อมูลหรือผลิตภัณฑ์ที่น่าสงสัย. (ตัวอย่างเช่น, การวิเคราะห์ของ Sokol ตั้งข้อสังเกตว่าการสอบเทียบที่ถูกลืมและชิ้นส่วนที่สึกหรอนั้นเกิดขึ้น “ความล้มเหลวง่ายๆ” ที่สามารถทำให้เกิดการระงับแบทช์ได้) รับรองบันทึกการบำรุงรักษาที่เข้มงวด, การใช้ตัวติดตามแบบดิจิทัลสำหรับกำหนดการสอบเทียบ, และการมอบอำนาจให้พนักงานแจ้งปัญหาอย่างรวดเร็วถือเป็นแนวทางปฏิบัติที่ดีที่สุดในการแก้ไขปัญหานี้.
- ปัญหาความสมบูรณ์ถูกต้องของข้อมูล: กลุ่มผลิตภัณฑ์ GMP สมัยใหม่อาศัยระบบอิเล็กทรอนิกส์. ความล้มเหลวในการควบคุมข้อมูลอาจทำให้ GxP เสียหายได้. ตัวอย่าง ได้แก่ เส้นทางการตรวจสอบที่ถูกปิดใช้งาน, รหัสผ่านที่อ่อนแอ, คัดลอก/วางข้อมูลแทนรายการต้นฉบับ, หรือความล้มเหลวในการตรวจสอบบันทึกทางอิเล็กทรอนิกส์. บริษัทต้องบังคับใช้หลักการ ALCOA+ — เช่น, ตรวจสอบให้แน่ใจว่าการป้อนข้อมูลทั้งหมดเป็น ระบุแหล่งที่มาได้ (เชื่อมโยงกับผู้ใช้), ชัดเจน (ชัดเจน), ร่วมสมัย (บันทึกแบบเรียลไทม์), ต้นฉบับ/ถูกต้อง, และ สมบูรณ์/สม่ำเสมอ. ฝึกอบรมผู้ปฏิบัติงานและระบบอัตโนมัติเมื่อเป็นไปได้ (E.G. บันทึกที่ล็อคด้วยคอมพิวเตอร์) ช่วยป้องกันการเขียนทับหรือการละเว้นด้วยตนเอง.
- การควบคุมการเปลี่ยนแปลงและข้อบกพร่อง CAPA: จำเป็นต้องมีกระบวนการจัดการการเปลี่ยนแปลงที่มีประสิทธิภาพสำหรับการเปลี่ยนแปลงใดๆ (การอัพเกรดอุปกรณ์, SOP ใหม่, วัตถุดิบใหม่). ข้อผิดพลาดทั่วไปคือการไม่บันทึกการเปลี่ยนแปลงหรือการข้ามการตรวจสอบความถูกต้องอีกครั้งหลังจากการแก้ไข. ในทำนองเดียวกัน, ความล้มเหลวในการตรวจสอบการเบี่ยงเบนอย่างเหมาะสม (การตัดปัญหาเป็นเพียง “ข้อผิดพลาดของมนุษย์” โดยไม่มีการวิเคราะห์สาเหตุที่แท้จริง) ปล่อยให้ปัญหายังคงอยู่ได้. หน่วยงานกำกับดูแลคาดหวัง CAPA ที่แข็งแกร่ง (การดำเนินการแก้ไขและป้องกัน) ระบบเพื่อแก้ไขความเบี่ยงเบนใด ๆ.
- ปัญหาการฝึกอบรมและวัฒนธรรม: GxP กำหนดให้บุคลากรทุกคนได้รับการฝึกอบรมและตระหนักถึงขั้นตอนด้านคุณภาพ. โปรแกรมการฝึกอบรมที่ไม่เพียงพอหรือการหมุนเวียนที่สูงอาจนำไปสู่การละเมิดโดยไม่ได้ตั้งใจ. การสร้างวัฒนธรรมที่มีคุณภาพ (โดยที่พนักงานรู้สึกว่าต้องรับผิดชอบต่อการปฏิบัติตามกฎระเบียบและได้รับการสนับสนุนให้รายงานปัญหา) มีความสำคัญแต่มักจะพัฒนาช้า.
โดยสรุป, ความท้าทายมักเป็นเรื่องขององค์กร: เอกสารประกอบ, การซ่อมบำรุง, การฝึกอบรม, และแนวปฏิบัติด้านข้อมูล. การเอาชนะสิ่งเหล่านี้หมายถึงการลงทุนในระบบ (เช่น ซอฟต์แวร์การจัดการเอกสารอิเล็กทรอนิกส์หรือการติดตามการสอบเทียบ), SOPs ที่มีระเบียบวินัย, และการตรวจสอบภายในบ่อยครั้ง. บริษัทที่ดำเนินการแก้ไขปัญหาเหล่านี้ในเชิงรุกจะพบว่าการปฏิบัติตาม GxP มีความราบรื่นมากขึ้นในระหว่างการตรวจสอบอย่างเป็นทางการ.
บริษัทยาบรรลุการปฏิบัติตามข้อกำหนด GxP ได้อย่างไร
การบรรลุการปฏิบัติตาม GxP คือ โครงการที่ครอบคลุมทั้งองค์กร. ด้านล่างนี้เป็นลำดับขั้นตอนทั่วไป (แสดงไว้ในผังงาน) ที่บริษัทยาปฏิบัติตามเพื่อสร้างระบบที่ปฏิบัติตามข้อกำหนด:
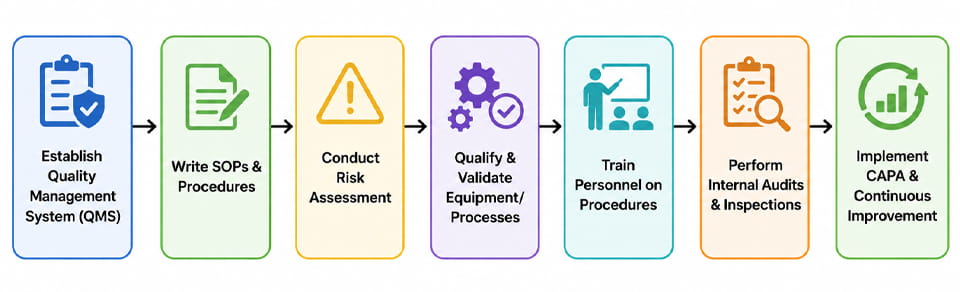
- ระบบการจัดการคุณภาพ (ระบบบริหารคุณภาพ): เริ่มต้นด้วยการกำหนดโครงสร้างองค์กรเพื่อคุณภาพ (E.G. คู่มือคุณภาพ, นโยบาย). ซึ่งรวมถึงการมอบหมายบทบาทและความรับผิดชอบที่มีคุณภาพ.
- พัฒนา SOP & เอกสาร: ร่างและอนุมัติขั้นตอนมาตรฐานในการผลิต, การทดสอบ, การควบคุมการเปลี่ยนแปลง, การเบี่ยงเบน, ฯลฯ. ตรวจสอบให้แน่ใจว่าแต่ละกระบวนการได้รับการบันทึกไว้อย่างชัดเจน.
- การประเมินความเสี่ยง: ดำเนินการประเมินความเสี่ยงอย่างเป็นทางการ (E.G. เอฟเอ็มอีเอ) เพื่อระบุพารามิเตอร์และการควบคุมกระบวนการที่สำคัญ. ข้อมูลนี้จะแจ้งว่าควรเน้นการตรวจสอบและติดตามตรงจุดใด.
- คุณสมบัติของอุปกรณ์/กระบวนการ (IR/WH/PQ): เช่นเดียวกับในส่วนที่แล้ว, ผ่านการรับรองอุปกรณ์และกระบวนการผลิตทั้งหมด. รักษาโปรโตคอลและรายงานการตรวจสอบโดยละเอียด.
- การฝึกอบรม: ผู้ประกอบการรถไฟ, วิศวกร, และเจ้าหน้าที่ QA/QC ในขั้นตอนที่ได้รับอนุมัติ, หลักการของ GxP, และการใช้อุปกรณ์.
- การตรวจสอบภายใน: ดำเนินการตรวจสอบตนเองเป็นประจำหรือจำลองการตรวจสอบเพื่อตรวจสอบความสม่ำเสมอและระบุปัญหา (E.G. ตรวจสอบบันทึกแบทช์, บันทึกสิ่งแวดล้อม, สถานะการสอบเทียบ).
- คาปา: เมื่อใดก็ตามที่มีการระบุการเบี่ยงเบนหรือการค้นพบ (ภายในหรือโดยหน่วยงานกำกับดูแล), ตรวจสอบสาเหตุที่แท้จริง, ใช้การดำเนินการแก้ไข, และปรับปรุงขั้นตอนเพื่อป้องกันการเกิดซ้ำ.
- การปรับปรุงอย่างต่อเนื่อง: ใช้ข้อมูล (E.G. การวิเคราะห์แนวโน้มจากรายงานการผลิตหรือบันทึกข้อร้องเรียน) เพื่อขับเคลื่อนการปรับปรุงคุณภาพและเพิ่มประสิทธิภาพกระบวนการ.
แต่ละขั้นตอนจะวนกลับตามความจำเป็น เช่น, การเปลี่ยนแปลงที่พบในระหว่างการตรวจสอบนำไปสู่การอัปเดต SOP และการฝึกอบรมใหม่. บริษัทต่างๆ ยังใช้การจัดการความเสี่ยงด้านคุณภาพ (QRM) และคุณภาพตามการออกแบบ (คิวบีดี) หลักการที่จะทำให้แนวทางนี้เป็นระบบ. ที่ ผังงานด้านบน แสดงให้เห็นถึงกระบวนการที่เป็นวัฏจักรนี้.
ความสัมพันธ์ระหว่าง GxP และอุปกรณ์บรรจุภัณฑ์
ทันสมัย เกี่ยวกับเภสัชกรรม สายการบรรจุ มีความซับซ้อนและต้องนำหลักการ GxP มาใช้โดยตรง. เครื่องจักรทุกเครื่องในสายการผลิต ตั้งแต่เครื่องถอดรหัสขวดไปจนถึงเครื่องบรรจุภัณฑ์ไปจนถึงเครื่องบรรจุกล่อง จะต้องได้รับการออกแบบและใช้งานในลักษณะที่ยึดถือ GMP. นี่คือการเชื่อมต่อที่สำคัญ:
- การออกแบบที่ถูกสุขลักษณะ: เครื่องบรรจุภัณฑ์ถูกสร้างขึ้นเพื่อหลีกเลี่ยงการปนเปื้อน. ตัวอย่างเช่น, อัน เครื่องบรรจุหีบห่อ จะมีส่วนที่ปิดล้อมและรางป้อนเรียบ, ผลิตภัณฑ์จึงไม่สัมผัสกับพื้นหรือพื้นผิวที่มีฝุ่น. เครื่องมือและชิ้นส่วนสำหรับบริเวณที่สัมผัสกับผลิตภัณฑ์มักเป็นสแตนเลสหรือพลาสติกเกรดทางการแพทย์, สอดคล้องกับข้อกำหนดวัสดุ GMP.
- การตรวจสอบ-พร้อมแล้ว: อุปกรณ์บรรจุภัณฑ์ต้องมีคุณสมบัติครบถ้วน. ซัพพลายเออร์มักออกแบบเครื่องจักรเพื่อให้ตรวจสอบได้ง่าย: พื้นที่ที่เข้าถึงได้สำหรับเซ็นเซอร์ (สำหรับการตรวจสอบการสอบเทียบ), ความสามารถในการวิ่งเปล่าและวิ่งเต็มที่, และประสิทธิภาพที่มั่นคง. ลูกค้าคาดหวังว่าเครื่องจักรจะมีข้อกำหนด (SOP และคู่มือ) ที่เชื่อมโยงโดยตรงกับกิจกรรม IQ/OQ/PQ.
- บันทึกแบทช์และการตรวจสอบย้อนกลับ: ทุกขั้นตอนในบรรทัด GMP ได้รับการบันทึกไว้. เครื่องบรรจุภัณฑ์อัตโนมัติมักจะทำงานร่วมกับซอฟต์แวร์เพื่อบันทึกหมายเลขแบทช์, ความเร็วของสาย, และเอาท์พุท. ตัวอย่างเช่น, สายการบรรจุขวดอาจติดฉลากแต่ละขวดโดยอัตโนมัติด้วยรหัสชุดและการประทับเวลา. รหัสเหล่านั้นเชื่อมโยงกลับไปยังการดำเนินการผลิต. ระบบยังสามารถบันทึกน้ำหนักหรือนับจำนวนการปฏิเสธได้อีกด้วย (E.G. แคปซูลที่บรรจุน้อยเกินไปถูกทำเครื่องหมายโดยเครื่องตรวจสอบน้ำหนักบนสายพาน). ข้อมูลนี้จะกลายเป็นส่วนหนึ่งของบันทึกชุดงานอิเล็กทรอนิกส์. ในระยะสั้น, เครื่องบรรจุภัณฑ์ช่วยบำรุงรักษา การตรวจสอบย้อนกลับ ของทุกหน่วย, ซึ่งเป็นข้อกำหนดด้านกฎระเบียบ.
- 21 ส่วนซีเอฟอาร์ 11 การปฏิบัติตาม: ตามที่กล่าวไว้ข้างต้น, การควบคุมด้วยคอมพิวเตอร์บนอุปกรณ์บรรจุภัณฑ์ (เช่น Human-Machine Interface และ PLC) ควรปฏิบัติตามกฎการบันทึกอิเล็กทรอนิกส์. ซึ่งหมายความว่าผู้ปฏิบัติงานเข้าสู่ระบบด้วย ID ที่ไม่ซ้ำกัน, และไม่มีการเปลี่ยนแปลงพารามิเตอร์ใด ๆ โดยไม่ได้รับอนุญาต. บันทึกข้อมูล (E.G. การตั้งค่า, ผลการทดสอบ) จะต้องปลอดภัยและมีการประทับเวลา. เครื่องจักรสมัยใหม่จำนวนมากในปัจจุบันมีระดับการเข้าถึงของผู้ใช้ด้วย (ผู้ปฏิบัติงานเทียบกับ. หัวหน้างาน) และคุณสมบัติบันทึกการตรวจสอบเพื่อตอบสนองความต้องการเหล่านี้.
- การลดข้อผิดพลาดและระบบอัตโนมัติ: การบรรจุอัตโนมัติช่วยลดการสัมผัสด้วยมือ, ซึ่งช่วยลดข้อผิดพลาดของมนุษย์ (โฟกัส GxP). เช่น, เครื่องเติมแคปซูล จาก Jinlu สามารถวิ่งด้วยความเร็วสูงพร้อมปริมาณที่แม่นยำ, ลดความจำเป็นในการแก้ไขด้วยตนเอง. เช่นเดียวกัน, เครื่องบรรจุกล่องอัตโนมัติ ให้แน่ใจว่ามีการปิดผนึกอย่างสม่ำเสมอ. เพื่อการปฏิบัติตาม, ซึ่งหมายความว่าโอกาสที่จะหยิบส่วนประกอบผิดหรือฉลากผิดน้อยลง ซึ่งสำคัญมากเมื่อบรรจุผลิตภัณฑ์ต่างๆ เคียงข้างกัน.
- คุณสมบัติด้านกฎระเบียบ: กฎระเบียบใหม่เช่น DSCSA (สหรัฐอเมริกา) หรือเอฟเอ็มดี (สหภาพยุโรป) ต้องการหมายเลขซีเรียลในแต่ละแพ็ค. เครื่องบรรจุภัณฑ์มักติดตั้งเครื่องพิมพ์โค้ด 2D และกล้องวิชันซิสเต็มเพื่อใช้/ตรวจสอบรหัสเหล่านี้. คุณสมบัติดังกล่าวแสดงให้เห็นว่ากฎหมายตลาดเป็นอย่างไร (ในนามของการตรวจสอบย้อนกลับของ GxP) ฟังก์ชั่นอุปกรณ์รูปร่าง.
- การสนับสนุนการปฏิบัติตามข้อกำหนด: อุปกรณ์ของ Jinlu, ตัวอย่างเช่น, พร้อมใช้งานตามมาตรฐาน GMP และมักมาพร้อมกับการสนับสนุนด้านคุณสมบัติและการตรวจสอบย้อนกลับ. ฟิลเลอร์แบบแคปซูลทั่วไปอาจมี CIP (ทำความสะอาดในสถานที่) ระบบและถังป้อนที่ถอดออกได้สำหรับการฆ่าเชื้อ. ก เส้นตุ่ม อาจรวมถึงประตูยามที่มีลูกโซ่นิรภัยเพื่อป้องกันการทำงานเมื่อเปิด. รายละเอียดการออกแบบเหล่านี้สนับสนุน GMP โดยตรง.
โดยเลือกใช้เครื่องจักรบรรจุภัณฑ์โดยคำนึงถึง GxP, บริษัทยาทำให้การปฏิบัติตามกฎระเบียบราบรื่นยิ่งขึ้น. เช่น, การติดตั้ง Jinlu เครื่องบรรจุแคปซูล หรือ เครื่องบรรจุกล่อง หมายความว่าผู้ซื้อมีเครื่องจักรที่สร้างขึ้นตามมาตรฐานยาอยู่แล้ว, พร้อมเอกสารประกอบ (เช่น FAT/SAT) พร้อมสำหรับการตรวจสอบ. ในที่สุด, อุปกรณ์ที่ได้รับการออกแบบมาอย่างดีถือเป็นหัวใจสำคัญของกระบวนการผลิตที่เป็นไปตามข้อกำหนด.

บทสรุป
GxP คือรากฐานของคุณภาพยา. มันไม่ใช่แค่ชุดของกฎเกณฑ์, แต่เป็นความมุ่งมั่นทั่วทั้งบริษัทในการสร้างความปลอดภัย, ยาที่มีประสิทธิภาพ. ที่แกนกลางของมัน, GxP รับประกันว่า “ยาถูกสร้างมาอย่างถูกต้อง” แนวปฏิบัติที่ดีในการผลิต (GMP) เป็นส่วนที่โดดเด่นที่สุดของ GxP สำหรับการผลิตยา, ครอบคลุมถึงการออกแบบอุปกรณ์สุขอนามัย, กระบวนการตรวจสอบแล้ว, และเอกสารที่เข้มงวด. ส่วนประกอบอื่นๆ เช่น GLP, จีซีพี, จีดีพี, และ GVP กล่าวถึงขั้นตอนต่างๆ (การศึกษาในห้องปฏิบัติการ, การทดลอง, การกระจาย, และการเฝ้าระวังด้านเภสัชกรรม, ตามลำดับ), แต่ทุกคนมีเป้าหมายในการปกป้องผู้ป่วยร่วมกัน.
อุปกรณ์บรรจุภัณฑ์ยา มีบทบาทสำคัญในการปฏิบัติตาม GxP. เครื่องจักรเช่นฟิลเลอร์แคปซูล, เครื่องบรรจุตุ่ม, และกล่องบรรจุจะต้องสร้างและตรวจสอบเพื่อให้ตรงตามมาตรฐาน GMP เป็นต้น, ทำความสะอาดง่าย, รองรับบันทึกชุดอิเล็กทรอนิกส์, และรักษาความสามารถในการสืบย้อน. โดยการเลือก เครื่องจักรพร้อมมาตรฐาน GMP และปฏิบัติตามระเบียบการด้านคุณสมบัติ (IR/WH/PQ), บริษัทต่างๆ สามารถบูรณาการหลักการ GxP เข้ากับสายการผลิตของตนได้.
พร้อมที่จะรับรองการปฏิบัติตาม GxP ในสายการผลิตของคุณ? พิจารณาอุปกรณ์บรรจุภัณฑ์ขั้นสูงของ Jinlu Packing, ซึ่งออกแบบมาสำหรับสภาพแวดล้อม GMP. ติดต่อจินลู่ เพื่อขอใบเสนอราคาหรือเรียนรู้เพิ่มเติมเกี่ยวกับเครื่องบรรจุแคปซูลที่ได้มาตรฐาน GMP ของเรา, เครื่องบรรจุตุ่ม, และเส้นบรรจุกล่อง.
คำถามที่พบบ่อยเกี่ยวกับ GxP ใน Pharma
GxP ย่อมาจากอะไร?
GxP ย่อมาจาก “แนวปฏิบัติที่ดี”. เป็นคำทั่วไปสำหรับหลักเกณฑ์ด้านคุณภาพต่างๆ ในด้านเภสัชกรรม. “x” สามารถเป็น M ได้ (การผลิต), ล (ห้องปฏิบัติการ), ค (คลินิก), ดี (การกระจาย), ฯลฯ. กล่าวอีกนัยหนึ่ง, แนวปฏิบัติที่ดีในการผลิต (GMP), แนวปฏิบัติที่ดีในห้องปฏิบัติการ (GLP), การปฏิบัติทางคลินิกที่ดี (จีซีพี), ฯลฯ.
GMP เป็นส่วนหนึ่งของ GxP หรือไม่?
ใช่. GMP (แนวปฏิบัติที่ดีในการผลิต) เป็นหนึ่งในองค์ประกอบหลักของ GxP. GxP คือร่ม, และ GMP หมายถึงการผลิตโดยเฉพาะ. ดังนั้นเมื่อเราพูดถึงการปฏิบัติตาม GxP, GMP มักเป็นจุดสนใจที่ใหญ่ที่สุดเนื่องจากควบคุมการผลิตและบรรจุภัณฑ์. ส่วนอื่นๆ เช่น GLP หรือ GVP ก็อยู่ในกลุ่มผลิตภัณฑ์ GxP เช่นกัน.
GxP และ GMP แตกต่างกันอย่างไร?
GxP เป็นหมวดหมู่กว้างๆ ของ “แนวทางปฏิบัติที่ดี” ทั้งหมดในเภสัชภัณฑ์ (ครอบคลุมการพัฒนาผ่านการจัดจำหน่าย). GMP เป็นเพียงหนึ่งในแนวทางปฏิบัติเหล่านั้น, มุ่งเน้นไปที่การผลิต. คิดว่า GxP เป็นกรอบงานคุณภาพทั้งหมด, และ GMP เป็นส่วนที่เกี่ยวข้องกับมาตรฐานการผลิต.
ใครเป็นผู้ควบคุมการปฏิบัติตาม GxP?
หน่วยงานกำกับดูแลบังคับใช้ GxP. ในประเทศสหรัฐอเมริกา, FDA ดูแล cGMP และ GLP, และ FDA/NIH จะดูแล GCP ในการทดลองทางคลินิก. ในยุโรป, EMA (และองค์กรระดับชาติเช่น MHRA) บังคับใช้หลักเกณฑ์ EU GMP และ GCP. WHO เผยแพร่แนวทาง GxP ระหว่างประเทศที่หลายประเทศนำมาใช้. แต่ละประเทศอาจมีเวอร์ชันของตัวเอง, แต่อย, EMA, WHO เป็นหน่วยงานหลักที่มีการอ้างอิงทั่วโลก.
คืออะไร 21 ส่วนซีเอฟอาร์ 11 และเหตุใดจึงสำคัญสำหรับ GxP?
21 ส่วนซีเอฟอาร์ 11 เป็นข้อบังคับของ FDA ของสหรัฐอเมริกาเกี่ยวกับบันทึกอิเล็กทรอนิกส์และลายเซ็นอิเล็กทรอนิกส์. สำหรับ GxP, มันหมายถึงระบบคอมพิวเตอร์ใด ๆ ที่สร้างบันทึก (เช่นซอฟต์แวร์ HMI หรือ LIMS ของเครื่อง) ต้องมีการควบคุมเพื่อให้บันทึกมีความปลอดภัย, ประทับเวลา, และไม่สามารถเปลี่ยนแปลงได้. ตัวอย่างเช่น, ระบบควบคุมของเครื่องบรรจุตุ่มจะต้องมีการเข้าสู่ระบบที่ปลอดภัยและเส้นทางการตรวจสอบสำหรับการเปลี่ยนแปลงพารามิเตอร์. การปฏิบัติตามส่วนหนึ่ง 11 เป็นข้อบังคับในสหรัฐอเมริกาและเป็นแนวทางในภูมิภาคอื่นๆ (ภาคผนวกของสหภาพยุโรป 11) บนระบบอิเล็กทรอนิกส์.
เครื่องบรรจุภัณฑ์ยาจำเป็นต้องปฏิบัติตาม GxP หรือไม่?
อย่างแน่นอน. อุปกรณ์ใดๆ ที่ใช้ในการผลิตหรือบรรจุภัณฑ์ยาต้องเป็นไปตามเกณฑ์การออกแบบและการตรวจสอบของ GxP. นี่หมายถึงเครื่องบรรจุภัณฑ์ (ฟิลเลอร์แคปซูล, เครื่องพุพอง, ฟิลเลอร์ของเหลว, กล่องกระดาษ, ฯลฯ) ควรมีการออกแบบที่ถูกสุขลักษณะ, การดำเนินการที่ได้รับการตรวจสอบแล้ว, และคุณสมบัติความสมบูรณ์ของข้อมูล. เช่น, สายการบรรจุต้องปฏิบัติตามขั้นตอนการทำความสะอาดที่บันทึกไว้ (ข้อกำหนด GMP) และใช้ระบบที่บันทึกข้อมูลแบทช์. ในหลายกรณี, ผู้จำหน่ายอุปกรณ์บรรจุภัณฑ์โฆษณาเครื่องจักรที่ "เป็นไปตามมาตรฐาน GMP" หรือ "พร้อมใช้โดย FDA". เมื่อเลือกอุปกรณ์, ผู้ผลิตต้องการคุณสมบัติต่างๆ เช่น ชิ้นส่วนหน้าสัมผัสที่ทำจากสแตนเลส, ทำความสะอาดง่าย, และโปรโตคอล IQ/OQ/PQ เต็มรูปแบบ. อุปกรณ์ของ Jinlu, ตัวอย่างเช่น, ได้รับการออกแบบโดยคำนึงถึงมาตรฐานเหล่านี้เพื่อรองรับสภาพแวดล้อมที่ควบคุมโดย GxP.
อ้างอิง:
1.แนวทางปฏิบัติที่ดีในการผลิต -- WHO
2.ตส 986 – ภาคผนวก 2: แนวปฏิบัติที่ดีในการผลิตผลิตภัณฑ์ยาของ WHO: หลักการสำคัญ -- WHO
3.แนวทางการผลิตยาของ WHO -- WHO
4.คำแนะนำเกี่ยวกับแนวทางปฏิบัติในการผลิตที่ดีและแนวทางปฏิบัติในการกระจายสินค้าที่ดี: คำถามและคำตอบ —— สำนักงานยาแห่งยุโรป
5.คำแนะนำ MHRA เกี่ยวกับความสมบูรณ์ถูกต้องของข้อมูล GxP —— gov.uk
6.คู่มือแนวปฏิบัติที่ดีของ GAMP: การทำงานของระบบคอมพิวเตอร์ GxP —— ispe.org




