W branży farmaceutycznej, Jakość i bezpieczeństwo pacjentów są najważniejsze. Każda tabletka, fiolka, lub opakowanie blistrowe musi być produkowane pod ścisłą kontrolą, aby zapewnić jego skuteczność, czysty, i bezpieczne. To właśnie tam Zgodność z GxP wchodzi. GxP to ogólny termin oznaczający „Dobry X Praktyka,”, gdzie „x” może oznaczać Produkcja, Laboratorium, Kliniczny, Dystrybucja, itp. Esencjonalnie, Wytyczne GxP mają charakter regulacyjny systemy jakości że firmy farmaceutyczne musi podążać dokumentować i kontrolować wszystkie procesy. Razem, zasady te zapewniają stałą produkcję leków i ich kontrolę zgodnie z wysokimi standardami jakości na każdym etapie rozwoju, produkcja, Testowanie, opakowanie, i dystrybucja.
Organy regulacyjne takie jak FDA (NAS. Administracja Żywności i Leków), EMA (Europejska Agencja Leków), KTO, a inne egzekwują standardy GxP. Na przykład, Aktualna dobra praktyka produkcyjna FDA (CGMP) zasady (21 Części CFR 210–211) ustalić minimalne wymagania dotyczące produkcji, uszczelka, oraz przechowywanie narkotyków, aby upewnić się, że są bezpieczne i mają odpowiedni skład i moc. Podobnie, Wytyczne WHO i UE obejmują GMP/GLP/GDP/GVP na całym świecie. W rzeczywistości, rozwijają się firmy farmaceutyczne Systemy Zarządzania Jakością oraz standardowe procedury operacyjne (standardowe procedury operacyjne) wokół zasad GxP. Każda partia produktu posiada identyfikowalne zapisy od początku do końca. Nawet sprzęt musi być kwalifikowany (z kwalifikacją instalacyjną/operacyjną/wydajnościową – IQ/OQ/PQ) i zatwierdzone pod kątem spełnienia wymagań GxP.

Co oznacza GxP w farmacji?
Akronim GxP oznacza Dobra praktyka „x”., gdzie "G" jest „Dobry” i "P" jest „Praktyka”. „x” oznacza różne dyscypliny. GxP nie jest pojedynczym rozporządzeniem, ale grupą wytycznych dotyczących jakości. Kluczowe dyscypliny GxP obejmują:
- Dobra Praktyka Produkcyjna (GMP): Zapewnia, że produkty są konsekwentnie produkowane i kontrolowane zgodnie ze standardami jakości. GMP obejmuje produkcję, sprzęt, udogodnienia, personel, i dokumentację w celu zminimalizowania skażenia, pomieszania, i błędy.
- Dobra Praktyka Laboratoryjna (GLP): Reguluje prowadzenie nieklinicznych badań laboratoryjnych (jak badania toksykologiczne) aby zapewnić integralność i odtwarzalność danych. GLP gwarantuje, że testy laboratoryjne są dobrze udokumentowane, kontrolowane, i niezawodne.
- Dobra praktyka kliniczna (GCP): Międzynarodowy standard projektowania, dyrygowanie, monitorowanie, i raportowanie badań klinicznych. GCP chroni prawa pacjentów, integralność danych, i zapewnia, że wyniki badań są wiarygodne.
- Dobra Praktyka Dystrybucyjna (PKB): Wyznacza standardy przechowywania, obsługiwanie, i transport produktów farmaceutycznych. Na przykład PKB zapewnia utrzymanie jakości leków w całym łańcuchu dostaw, poprzez kontrolowanie temperatury, unikanie zanieczyszczeń, i śledzenie partii.
- Dobra praktyka nadzoru nad bezpieczeństwem farmakoterapii (Partner partnerski): Zawiera wytyczne dotyczące monitorowania bezpieczeństwa leków i zgłaszania zdarzeń niepożądanych po wprowadzeniu produktu na rynek. GVP pomaga firmom farmaceutycznym i organom regulacyjnym wykrywać, oceniać, i zapobiegać negatywnym skutkom ubocznym.
Każdy obszar GxP jest egzekwowany przez określone przepisy lub wytyczne (NP., FDA 21 CFR dla GMP/GLP/GCP, EU EudraLex dla GMP/GDP/GVP, I wytyczne, KTO, itp.). Chociaż szczegóły się różnią, wspólnym celem jest aby mieć pewność, że produkty lecznicze są bezpieczne, skuteczny, i wysoką jakość na każdym kroku.
Tabela: Główne dyscypliny GxP(Rodzaje przepisów GxP)
| Typ GxP |
Pełne imię i nazwisko |
Etap cyklu życia produktu |
Cel/cel |
| GLP |
Dobra Praktyka Laboratoryjna |
Badania leków |
Reguluje niekliniczne badania laboratoryjne (jak testy toksyczności) aby zapewnić jakość i identyfikowalność danych. GLP wyznacza standardy procedur laboratoryjnych, rejestracja danych, i raportowanie. |
| GCP |
Dobra Praktyka Laboratoryjna |
Badania kliniczne |
Międzynarodowe standardy badań klinicznych (I-GCP). Obejmuje projekt próbny, świadoma zgoda, monitorowanie, i dokładne raportowanie wyników. |
| GMP |
Dobra praktyka kliniczna |
Produkcja |
Zapewnia, że leki są konsekwentnie wytwarzane i kontrolowane podczas produkcji/opakowania. Obejmuje standardy obiektu/wyposażenia, Szkolenie personelu, Testowanie kontroli jakości. |
| PKB |
Dobra Praktyka Dystrybucyjna |
Magazynowanie & Logistyka |
Zapewnia właściwe przechowywanie i transport leków (NP., prawidłowa temperatura, bezpieczna obsługa) dzięki czemu jakość jest utrzymywana w całym łańcuchu dostaw. |
| Partner partnerski |
Dobra praktyka nadzoru nad bezpieczeństwem farmakoterapii |
Monitorowanie po wprowadzeniu na rynek |
Wytyczne dotyczące bieżącego monitorowania bezpieczeństwa leków dostępnych na rynku (zgłaszanie zdarzeń niepożądanych, zarządzanie ryzykiem, komunikacja z władzami). |
Dlaczego GxP jest ważny w przemyśle farmaceutycznym?
Zgodność z GxP jest niezbędna, ponieważ życie dosłownie od tego zależy. Jego głównymi celami są:
- Bezpieczeństwo pacjenta: Przestrzeganie GxP minimalizuje ryzyko, takie jak zanieczyszczenie, pomieszania, lub błędy w dozowaniu. Na przykład, Zasady GMP (jak właściwa higiena i kontrola procesu) pomagają zapobiegać zanieczyszczeniu krzyżowemu pomiędzy produktami. Bez GxP, Leki niespełniające norm mogą zaszkodzić pacjentom lub okazać się nieskuteczne. Organy regulacyjne podkreślają to „Zasady GxP istnieją z jednego powodu: chronić pacjentów”.
- Jakość i spójność produktu: Ramy GxP zapewniają, że każda partia leku spełnia te same specyfikacje. Obejmuje to stałą moc, czystość, i stabilność. Egzekwując rygorystyczne kontrole (np. kalibracja przyrządów, sprawdzone metody, testowanie w trakcie procesu), producenci mogą zagwarantować, że każda fiolka lub tabletka jest zgodna z informacjami na etykiecie.
- Zgodność z przepisami i zaufanie: Przestrzeganie zasad GxP jest prawnie obowiązkowe. Agencje takie jak FDA, EMA, MHRA, PMDA, i WHO polegają na inspekcjach i audytach GxP. Firmy, które wykazują silne praktyki GxP, budują zaufanie wśród organów regulacyjnych. Nieprzestrzeganie może skutkować wysłaniem ostrzeżeń, kary, wycofanie produktu, lub gorzej. Jak zauważa jeden z przewodników branżowych, organy regulacyjne przeprowadzają audyty w celu „weryfikacji zgodności ze standardami GxP”, co podkreśla integralność produktu i bezpieczeństwo pacjentów.
- Integralność danych: Krytycznym aspektem GxP jest zapewnienie dokładności i wiarygodności danych. Nowoczesne regulacje (np. FDA 21 Część CFR 11, Załącznik UE 11) wymagają, aby zapisy elektroniczne były bezpieczne, oznaczone czasem, i odporne na manipulacje. Kluczowe zasady, takie jak ALCOA+ (Można przypisać, Czytelny, Współczesny, Oryginalny, Dokładny, plus Kompletne, Spójny, Cierpliwy, Dostępny) służą do oceny jakości danych. W rzeczywistości, oznacza to wdrożenie ścieżek audytu w oprogramowaniu, blokowanie dokumentów głównych, oraz regularne przeglądanie zapisów partii. Solidna integralność danych jest podstawą GxP; bez tego, nawet dobrze wykonanym produktom nie można ufać.
Krótko mówiąc, GxP jest podstawą zarządzania jakością w branży farmaceutycznej. Zapewnia to każdy krok — od testów laboratoryjnych po końcowe pakowanie — odbywa się pod kontrolą, udokumentowane warunki. Na przykład, jeden z przewodników to zauważa „Zgodność z GxP gwarantuje opracowywanie leków i środków biologicznych, zrobiony fabrycznie, i testowane zgodnie z rygorystycznymi normami,” zapobieganie kosztownym zanieczyszczeniom lub błędom w etykietowaniu. Zatem, GxP nie tylko chroni pacjentów, ale także wspiera wiarygodną reputację marki i dostęp do rynku dla firm farmaceutycznych.

Zrozumienie GMP – najważniejszego standardu GxP
Chociaż wszystkie obszary GxP są krytyczne, GMP (Dobra Praktyka Produkcyjna) jest często postrzegane jako podstawa zgodności produkcji farmaceutycznej. GMP obejmuje cały proces wytwarzania produktów leczniczych, łącznie z surowcami, sprzęt, udogodnienia, procesy, i linie pakujące. Jego głównym celem jest zminimalizować ryzyko zanieczyszczenia produktu lub odstępstw poprzez egzekwowanie rygorystycznych kontroli i dokumentacji. Kluczowe elementy GMP obejmują:
- Projekt obiektu i wyposażenia: Sprzęt powinien być zaprojektowany z myślą o higienicznej pracy. Oznacza to użycie materiałów klasy farmaceutycznej (np. 316Stal nierdzewna), gładkie spoiny, brak martwych stref, i łatwy dostęp do czyszczenia. Do maszyn pakujących, może to obejmować przenośniki wspornikowe, części szybkozłączne, i ogrodzeni strażnicy. Właściwy układ zapobiega pomyłkom i ułatwia walidację czyszczenia (zatwierdzone procedury czyszczenia).
- Narzędzia i kalibracja: Wszystkie media (woda, sprężone powietrze, elektryczny) musi spełniać wymagania jakościowe. Przyrządy i czujniki (waga, przepływomierze, sondy temperatury) wymagają regularności kalibrowanie zgodnie z pisemnymi procedurami. FDA wyraźnie oczekuje zapisów kalibracji dla sprzętu krytycznego. Na przykład, pompa wolumetryczna linii napełnionych butelek byłaby skalibrowana, aby zapewnić dokładność każdej dawki.
- Walidacja i kwalifikacja: GMP wymaga, aby sprzęt i procesy były kwalifikowane i walidowane. Obejmuje to kwalifikację instalacyjną (ILORAZ INTELIGENCJI), Kwalifikacja operacyjna (OK), i Kwalifikacja Wykonawcza (PQ) aby udowodnić, że system działa zgodnie z przeznaczeniem (szczegółowo omówione poniżej). Na przykład, nowa maszyna pakująca w blistry musi zostać przetestowana pod kątem IQ/OQ/PQ, aby potwierdzić temperaturę zgrzewania, integralność uszczelnienia, i system indeksowania spełniają specyfikacje. Każda krytyczna maszyna (Wypełniacze kapsułki, kartoniarze, linie do napełniania płynem) muszą przestrzegać tego cyklu życia walidacji.
- Standardowe procedury operacyjne (standardowe procedury operacyjne): Szczegółowy, pisemne SOP są obowiązkowe na etapach produkcyjnych, czyszczenie, konserwacja, i kontroli jakości. Operatorzy muszą zostać przeszkoleni w zakresie tych standardowych procedur operacyjnych, a wszelkie zmiany muszą być kontrolowane poprzez formalny system kontroli zmian. Audytorzy szukają aktualnych SOP i zapisów szkoleniowych.
- Dobre praktyki dokumentacyjne: GMP to podkreśla „jeśli nie jest to udokumentowane, to się nie wydarzyło.” Wszystkie procedury, zapisy partii, czyszczenie dzienników, oraz testy kontroli jakości muszą być rejestrowane w sposób czytelny i jednocześnie. Wypełniony rejestr produkcji seryjnej (BMR) musi obejmować uzgodnienie materiałów, ustawienia sprzętu, kontrole w trakcie procesu, oraz wszelkie odchylenia/korekty. Współczesne trendy wymuszają elektroniczne zapisy wsadowe (eBR) ze ścieżkami audytu poniżej 21 Część CFR 11.
- Kontrola jakości (Kontrola jakości) i wydanie partii: Testowanie kontroli jakości (np. moc, sterylność, identyfikacja) odbywa się na surowcach i wyrobach gotowych. Partia zostaje zwolniona dopiero po przejrzeniu całej dokumentacji i wyników testów przez działy QA/QC. Ta końcowa kontrola jest kluczową częścią GMP.
Blog Jinlu Packing na temat opakowań GMP podkreśla kilka z tych wymagań. Na przykład, zauważa to konserwacja zapobiegawcza i kalibracja są wymagane przez GMP (21 CFR 211.68 wymaga określonych odstępów czasu i zapisów). To także podkreśla automatyzacja & zapobieganie błędom: nowoczesne linie pakujące GMP wykorzystują kontrolę wizyjną, skanowanie kodów kreskowych, i blokady, aby uniknąć błędów ludzkich. Integralność danych i kontrola komputera są również objęte GMP: wszystkie skomputeryzowane systemy na linii (np. sterowniki PLC, systemy wizyjne, Hmis) musi spełniać 21 Część CFR 11 – czyli unikalne loginy, ścieżki audytu, podpisy elektroniczne, i bezpieczne prowadzenie rejestrów. W rzeczywistości, oznacza to, że oprogramowanie sterujące maszyny będzie rejestrować każdą zmianę parametrów ze znacznikiem czasu i identyfikatorem użytkownika, i będzie wymagał podpisu elektronicznego menedżera w celu zatwierdzenia partii.
Lista kontrolna GMP dla sprzętu pakującego
Pomaga przeglądać wymagania GMP w formie listy kontrolnej. Do maszyn pakujących, producenci zazwyczaj zapewniają:
- Projekt & Przybory: Higieniczna konstrukcja (części ze stali nierdzewnej, Uszczelki zatwierdzone przez FDA), brak „martwych” obszarów, pochyłe powierzchnie, łatwy demontaż w celu czyszczenia.
- Walidacja: Ukończ IQ/OQ/PQ na każdej maszynie. Udokumentowane testowanie funkcji (opieczętowanie, pożywny, ważenie) i wydajność. (Zobacz sekcję Kwalifikacja sprzętu poniżej.)
- Czyszczenie: Zatwierdzone procedury czyszczenia za pomocą testów wymazowych lub płukania (zazwyczaj przy użyciu TOC lub specjalnych testów) i zapisy.
- Konserwacja & Kalibrowanie: Harmonogramy konserwacji zapobiegawczej z dziennikami. Kalibracja pomp dozujących, waga, czujniki zapewniające dokładność.
- Sterownica & Automatyzacja: Systemy kontroli wizyjnej (NP., wykrywacze metali, wagi kontrolne) w celu wykrycia usterek, czujniki zapobiegające nieprawidłowemu podawaniu, blokady zatrzymujące się w przypadku otwarcia drzwi.
- Integralność danych: 21 Część CFR 11 zgodność – ścieżki audytu, kontrola dostępu użytkowników, podpisy elektroniczne, zabezpieczone dane. Wszystkie rekordy (standardowe procedury operacyjne, dane wsadowe, odchylenia) przechowywane w stanie niezmienionym.
- Identyfikowalność: Obsługa serializacji/UID, kod kreskowy, łączenie każdego opakowania podstawowego z zapisami partii, umożliwienie wycofania w razie potrzeby. (Maszyny Jinlu często integrują drukarki etykiet lub czytniki kodów w celu zapewnienia identyfikowalności.)
- Środowiskowy & Sterowanie linią: W razie potrzeby odpowiednie standardy pomieszczeń czystych, udokumentowane kontrole prześwitu linii pomiędzy partiami, właściwe etykietowanie i obchodzenie się z materiałami, aby zapobiec pomyłkom.
Przestrzegając tych kontroli, producenci mogą być pewni, że ich urządzenia pakujące będą działać zgodnie z wytycznymi GMP. Na przykład, maszyny do napełniania kapsułki I Maszyny pakowania pęcherzy firmy Jinlu są zbudowane z funkcjami gotowymi na GMP (gładkie powierzchnie GMP, Możliwość CIP, itp.) aby sprostać tym wymaganiom.

Wymagania GxP dla producentów sprzętu farmaceutycznego
Dostawcy sprzętu pakującego odgrywają kluczową rolę w GxP. Kupujący oczekują maszyn, które będą nie tylko solidne i wydajne, ale także zbudowany w celu ułatwienia zgodności z przepisami. Kluczowe wymagania obejmują:
- Wsparcie kwalifikacyjne (IR/WH/PQ): Sprzedawcy powinni zapewnić protokoły i pomoc w kwalifikacjach. Oznacza to udokumentowaną kwalifikację instalacyjną (ILORAZ INTELIGENCJI) aby pokazać, że maszyna została zainstalowana prawidłowo, Kwalifikacja operacyjna (OK) aby udowodnić, że działa zgodnie ze specyfikacją, i Kwalifikacja Wykonawcza (PQ) aby sprawdzić, czy konsekwentnie zapewnia akceptowalny wynik. Na przykład, Jinlu Packing dostarcza każdą maszynę z zestawem IQ/OQ/PQ i pełnymi protokołami FAT/SAT. Szablony te często mogą zostać dostosowane przez zespół ds. kontroli jakości kupującego, oszczędność czasu podczas walidacji. Dostawca może również uczestniczyć w przebiegach kwalifikacyjnych lub udostępniać listy kontrolne certyfikowanego sprzętu.
- Pakiet Dokumentacji: Razem z maszyną, dostawcy powinni dostarczyć komplet dokumentów. Typowe elementy obejmują Instrukcja obsługi, Główna lista części, Schematy elektryczne, I Instrukcje konserwacji. Co ważne, TŁUSZCZ (Fabryczny test akceptacyjny) I SOBOTA (Test akceptacji witryny) raporty dokumentują, że maszyna przeszła testy fabryczne i na miejscu. Do każdego urządzenia pomiarowego należy dołączyć świadectwa wzorcowania. W rzeczywistości, zgodny pakiet dokumentacji będzie również zawierał specyfikacje systemu, SOP czyszczenia, oceny ryzyka, oraz jakakolwiek historia kontroli zmian.
- Funkcje integralności danych: Nowoczesny sprzęt powinien oferować elektroniczne elementy sterujące zgodne ze standardami danych GxP. Obejmuje to bezpieczne konta użytkowników (dostęp oparty na rolach), obowiązkowe podpisy elektroniczne w przypadku działań krytycznych, oraz pełne ścieżki audytu wszelkich zmian parametrów. Na przykład, ekran HMI może wymagać zalogowania się kierownika zmiany, aby rozpocząć produkcję, a każda zmiana przepisu lub ustawienia jest oznaczona znacznikiem czasu. Maszyny mogą również obsługiwać cyfrowe wyjście rekordów wsadowych, integracja z systemami MES/ERP.
- Narzędzia do walidacji i testowania: Niektórzy dostawcy udostępniają narzędzia programowe do rejestrowania danych, kalibrowanie, lub walidacja. Może to być preinstalowane oprogramowanie do przeprowadzania testów czujników, lub wbudowana funkcja blokowania parametrów podczas przebiegów PQ. Funkcje te zmniejszają wysiłek ręczny podczas walidacji.
- Projekt higieniczny i bezpieczeństwa: Sprzęt musi być łatwy do czyszczenia i konserwacji. Funkcje takie jak części szybkozłączne, strefy bezproduktowe, i CIP (Czyszczenie na miejscu) Opcje pomagają spełnić wymagania dotyczące czyszczenia. Materiały mające kontakt z produktem powinny być obojętne (np. 316L SS, Tworzywa sztuczne zatwierdzone przez FDA). Osłony zabezpieczające i blokady chronią operatorów, ale także zapewnić zgodność (np. maszyna zatrzymuje się po otwarciu).
- Wsparcie po sprzedaży: Zgodność z GxP jest w toku. Producenci mogą wymagać okresowej ponownej kwalifikacji lub ponownej kalibracji. Dostawcy powinni oferować usługi w cyklu życia: części zapasowe (do szybkiej wymiany zatwierdzonych części), umowy serwisowe, oraz aktualizacje dokumentacji walidacyjnej w przypadku wystąpienia zmian. Gotowość dostawcy do świadczenia usług kwalifikacyjnych na miejscu (IR/WH/PQ) może znacznie ułatwić działania mające na celu zapewnienie zgodności.
Lista kontrolna kwalifikacji sprzętu: Poniższa tabela podsumowuje typowe kroki i dokumenty wymagane do kwalifikacji nowej maszyny farmaceutycznej:
| Faza |
Kluczowe działania |
Typowe dokumenty |
| Wymagania użytkownika (URS) |
Zdefiniuj specyfikacje krytyczne (np. szybkość wyjściowa, dokładność) |
Specyfikacja wymagań użytkownika |
| Kwalifikacja projektowa (DQ) |
Sprawdź, czy projekt dostawcy jest zgodny z URS |
Raport z przeglądu specyfikacji projektu |
| Akceptacja fabryczna (TŁUSZCZ) |
Testy fabryczne głównych funkcji, często odzwierciedlają testy IQ/OQ |
Raport FAT |
| Kwalifikacja instalacyjna (ILORAZ INTELIGENCJI) |
Potwierdź poprawną instalację: narzędzia, konfiguracja mechaniczna, dokumentacja (rysunki, certyfikaty) |
Protokół IQ & lista kontrolna |
| Kwalifikacja operacyjna (OK) |
Przetestuj wszystkie funkcje: wydajność na pustym biegu, sterownica, alarmy, funkcje bezpieczeństwa |
Protokół OQ & wyniki |
| Kwalifikacja wydajności (PQ) |
Uruchom pełną produkcję z prawdziwym produktem: sprawdź jakość wyjściową, konsystencja, warunki stresowe |
Protokół PQ, prowadzić zapisy, przykładowe wyniki testów |
| Wersja ostateczna |
Przejrzyj wszystkie zapisy dotyczące kwalifikacji; Zatwierdzenie kontroli jakości w celu wprowadzenia maszyny do produkcji GMP |
Raport podsumowujący kwalifikacje |
(Notatka: Maszyny Jinlu są dostarczane z Szablony IQ/OQ/PQ i pełny Akceptacja fabryczna pakiet dokumentacji, które klienci mogą dostosować w zależności od potrzeb.) To ustrukturyzowane podejście – od wymagań użytkownika po PQ – jest wymagane przez przepisy takie jak Załącznik GMP UE 15 i wytyczne FDA. Pominięcie dowolnego kroku może spowodować lukę w przestrzeganiu przepisów.

Typowe wyzwania związane ze zgodnością z GxP
Nawet przy jasnych zasadach, firmy często napotykają przeszkody w przestrzeganiu zasad GxP. Niektóre częste wyzwania obejmują:
- Niekompletna walidacja lub dokumentacja: Jedną z najpoważniejszych luk jest używanie sprzętu bez pełnych zapisów IQ/OQ/PQ. „Pomijanie weryfikacji,” lub mieć częściowy kwalifikacja, jest uważany za „fatalna luka w GMP”. Podobnie, brakujące lub niedbałe zapisy partii i SOP podważają zgodność: inspektorzy są przeszkoleni w zakresie wyszukiwania dokumentacji. Jak to ujął jeden z ekspertów: „Jeśli nie jest to zapisane, to się nie wydarzyło.” Słabe prowadzenie rejestrów (utracone pliki, nieczytelne notatki, przestarzałe wersje) to powszechna czerwona flaga.
- Brak konserwacji i kalibracji sprzętu: Organy regulacyjne wielokrotnie stwierdzają problemy, takie jak przeterminowane terminy kalibracji lub opóźnione naprawy. Uszkodzony czujnik lub nieskalibrowana skala prowadzą do podejrzanych danych lub produktu. (Na przykład, Z analizy Sokola wynika, że zapomniane kalibracje i zużyte części są „proste awarie” co może spowodować wstrzymanie partii.) Zapewnienie ścisłych dzienników konserwacji, wykorzystanie cyfrowych trackerów do harmonogramów kalibracji, najlepszymi praktykami pozwalającymi przezwyciężyć ten problem jest umożliwienie pracownikom szybkiego sygnalizowania problemów.
- Problemy z integralnością danych: Nowoczesne linie GMP opierają się na systemach elektronicznych. Błędy w kontroli danych mogą złamać GxP. Przykładami są wyłączone ścieżki audytu, słabe hasła, skopiowane/wklejone dane zamiast oryginalnych wpisów, lub brak przeglądu dzienników elektronicznych. Firmy muszą egzekwować zasady ALCOA+ – m.in., upewniając się, że wszystkie wpisy danych są Można przypisać (powiązany z użytkownikiem), Czytelny (jasne), Współczesny (nagrywane w czasie rzeczywistym), Oryginał/dokładny, I Kompletny/spójny. Szkolenie operatorów i automatyzacja tam, gdzie to możliwe (np. zapisy zablokowane komputerowo) pomagają zapobiegać ręcznemu nadpisywaniu lub pominięciu.
- Kontrola zmian i niedobory CAPA: Wszelkie zmiany wymagają solidnego procesu zarządzania zmianami (ulepszenia sprzętu, nowe SPO, nowe surowce). Częstym błędem jest nieudokumentowanie zmiany lub pominięcie ponownej walidacji po modyfikacji. Podobnie, brak odpowiedniego zbadania odchyleń (spisywanie problemów jako zwykłego „błędu ludzkiego” bez analizy przyczyn źródłowych) może pozwolić, aby problemy nadal występowały. Organy regulacyjne oczekują silnego CAPA (działania korygujące i zapobiegawcze) systemów w celu wyeliminowania wszelkich odchyleń.
- Kwestie szkoleniowe i kulturalne: GxP wymaga, aby cały personel był przeszkolony i świadomy procedur jakości. Nieodpowiednie programy szkoleniowe lub duża rotacja mogą prowadzić do niezamierzonych naruszeń. Budowanie kultury jakości (gdzie pracownicy czują się odpowiedzialni za zgodność i są zachęcani do zgłaszania problemów) jest niezbędna, ale często rozwija się powoli.
Podsumowując, wyzwania mają często charakter organizacyjny: dokumentacja, konserwacja, szkolenie, i praktyki dotyczące danych. Pokonanie ich oznacza inwestycję w systemy (jak oprogramowanie do elektronicznego zarządzania dokumentami lub oprogramowanie do śledzenia kalibracji), zdyscyplinowane SOP, oraz częste audyty wewnętrzne. Firmy, które aktywnie zajmują się tymi obszarami, podczas oficjalnych inspekcji stwierdzą, że zgodność z GxP jest łatwiejsza.
Jak firmy farmaceutyczne osiągają zgodność z GxP
Osiągnięcie zgodności z GxP to a projekt obejmujący całą organizację. Poniżej znajduje się typowa sekwencja kroków (zilustrowano na schemacie blokowym) którymi kieruje się firma farmaceutyczna, aby zbudować zgodny system:
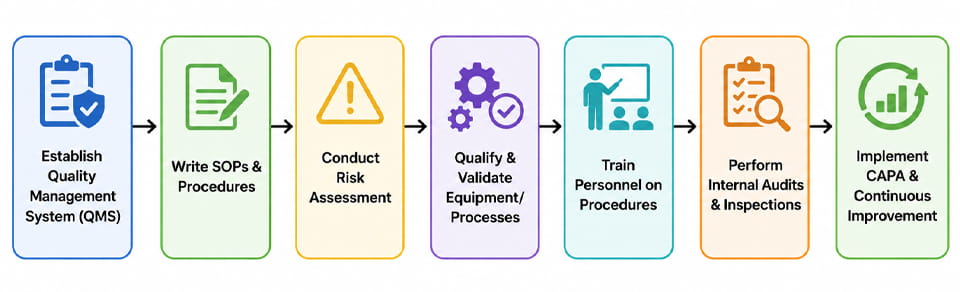
- System Zarządzania Jakością (Zarządzanie jakością): Zacznij od zdefiniowania struktury organizacyjnej ds. jakości (np. podręcznik jakości, polityki). Obejmuje to przypisywanie ról i obowiązków związanych z jakością.
- Opracuj standardowe procedury operacyjne & Dokumentacja: Opracuj i zatwierdź standardowe procedury produkcyjne, Testowanie, zmienić kontrolę, odchylenia, itp. Upewnij się, że każdy proces jest jasno udokumentowany.
- Ocena ryzyka: Przeprowadzaj formalną ocenę ryzyka (np. FMEA) w celu identyfikacji krytycznych parametrów procesu i kontroli. Informuje to, gdzie należy skoncentrować się na walidacji i monitorowaniu.
- Kwalifikacja sprzętu/procesu (IR/WH/PQ): Podobnie jak w poprzedniej części, kwalifikować cały sprzęt i procesy produkcyjne. Utrzymuj szczegółowe protokoły i raporty z walidacji.
- Szkolenie: Operatorzy pociągów, inżynierowie, oraz personel QA/QC na temat zatwierdzonych procedur, zasady GxP, i użytkowanie sprzętu.
- Audyty wewnętrzne: Przeprowadzaj rutynowe samokontrole lub pozorowane audyty, aby sprawdzić przestrzeganie zasad i wykryć problemy (np. sprawdź zapisy partii, dzienniki środowiskowe, stan kalibracji).
- CAPA: Za każdym razem, gdy zostanie zidentyfikowane odchylenie lub ustalenie (wewnętrznie lub przez regulator), zbadać pierwotną przyczynę, zastosować działania korygujące, i zaktualizować procedury, aby zapobiec ponownemu wystąpieniu.
- Ciągłe doskonalenie: Wykorzystaj dane (np. analiza trendów na podstawie raportów produkcyjnych lub dzienników reklamacji) w celu poprawy jakości i optymalizacji procesów.
Każdy krok zapętla się w razie potrzeby – np., zmiana wykryta podczas audytu prowadzi do aktualizacji SOP i przekwalifikowania. Firmy korzystają również z Zarządzania Ryzykiem Jakości (QRM) i Jakość według projektu (QbD) zasady umożliwiające usystematyzowanie tego podejścia. The schemat blokowy powyżej ilustruje ten cykliczny proces.
Związek między GxP a urządzeniami pakującymi
Nowoczesny farmaceutyczny Linie pakowania są złożone i muszą bezpośrednio odzwierciedlać zasady GxP. Każda maszyna na linii – od rozszyfrowywaczy butelek, przez maszyny blistrowe, po kartoniarkę – musi być zaprojektowana i używana w sposób zgodny z GMP. Oto najważniejsze połączenia:
- Higieniczny projekt: Maszyny pakujące są zbudowane tak, aby uniknąć zanieczyszczeń. Na przykład, A Blister Packaging Maszyna będzie miał zamkniętą sekcję formowania i gładkie tory podawania, aby produkt nie miał kontaktu z podłogą ani zakurzonymi powierzchniami. Narzędzia i części przeznaczone do obszarów mających kontakt z produktem są zazwyczaj wykonane ze stali nierdzewnej lub tworzywa sztucznego klasy medycznej, dostosowanie się do wymagań materiałowych GMP.
- Gotowy do walidacji: Sprzęt pakujący musi być w pełni kwalifikowany. Dostawcy często projektują maszyny w celu łatwej walidacji: dostępne obszary dla czujników (do kontroli kalibracji), możliwość wykonywania pustych i pełnych przebiegów, i stabilna wydajność. Klienci oczekują, że maszyny będą miały specyfikacje (SOP i podręczniki) które wiążą się bezpośrednio z działaniami IQ/OQ/PQ.
- Zapisy partii i identyfikowalność: Każdy krok na linii GMP jest dokumentowany. Zautomatyzowane maszyny pakujące często integrują się z oprogramowaniem do rejestrowania numerów partii, prędkości linii, i wyjście. Na przykład, linia rozlewnicza może automatycznie oznaczyć każdą butelkę kodem partii i znacznikiem czasu. Kody te są powiązane z serią produkcyjną. Systemy mogą również rejestrować wagę lub liczyć odrzuty (np. kapsułki z niedostatecznym wypełnieniem, oznaczone przez wagę kontrolną in-line). Dane te stają się częścią elektronicznego rejestru partii. Krótko mówiąc, maszyny pakujące pomagają w utrzymaniu identyfikowalność każdej jednostki, co jest wymogiem regulacyjnym.
- 21 Część CFR 11 Zgodność: Jak wspomniano wcześniej, skomputeryzowane kontrole urządzeń pakujących (jak interfejs człowiek-maszyna i sterownik PLC) powinny przestrzegać zasad prowadzenia ewidencji elektronicznej. Oznacza to, że operatorzy logują się przy użyciu unikalnych identyfikatorów, i żadne parametry nie mogą być zmieniane bez autoryzacji. Dzienniki danych (np. ustawienia, wyniki testów) muszą być bezpieczne i opatrzone datą. Wiele nowoczesnych maszyn zawiera obecnie poziomy dostępu użytkownika (operator vs. kierownik) oraz funkcje dziennika audytu spełniające te potrzeby.
- Redukcja błędów i automatyzacja: Automatyczne pakowanie ogranicza konieczność ręcznego dotykania, co ogranicza błędy ludzkie (skupienie się na GxP). Na przykład, maszyny do napełniania kapsułki firmy Jinlu może pracować z dużą prędkością przy precyzyjnym dozowaniu, minimalizując potrzebę ręcznej korekty. Podobnie, automatyczne kartoniarki zapewnić spójne uszczelnienie. Dla zgodności, oznacza to mniejsze ryzyko źle dobranych komponentów lub błędnych etykiet – co ma kluczowe znaczenie w przypadku pakowania różnych produktów obok siebie.
- Funkcje regulacyjne: Nowe regulacje jak DSCSA (USA) lub FMD (UE) żądać numerów seryjnych na poszczególnych opakowaniach. Maszyny pakujące są często wyposażone w drukarki kodów 2D i kamery wizyjne do nanoszenia/weryfikacji tych kodów. Takie cechy pokazują, jak działają prawa rynkowe (w imię identyfikowalności GxP) kształtować funkcje sprzętu.
- Wsparcie zgodności: Sprzęt Jinlu, Na przykład, jest zgodny z GMP i często zapewnia wsparcie w zakresie kwalifikacji i identyfikowalności. Typowy wypełniacz kapsułek może być wyposażony w CIP (czyszczenie na miejscu) systemem i wyjmowanym zbiornikiem na paszę do sterylizacji. A linia pęcherzy mogą obejmować drzwi ochronne z blokadami bezpieczeństwa uniemożliwiającymi działanie po otwarciu. Te szczegóły projektu bezpośrednio wspierają GMP.
Wybierając maszyny pakujące z myślą o GxP, firmy farmaceutyczne ułatwiają przestrzeganie przepisów. Na przykład, instalowanie Jinlu maszyna do napełniania kapsułek Lub maszyna kartonowa oznacza, że kupujący ma już maszynę zbudowaną zgodnie ze standardami farmaceutycznymi, z dokumentacją (jak FAT/SAT) gotowy do walidacji. Ostatecznie, dobrze zaprojektowany sprzęt jest podstawą zgodnego procesu produkcyjnego.

Wniosek
GxP jest podstawą jakości farmaceutycznej. To nie tylko zbiór zasad, ale zobowiązanie całej firmy do zapewnienia bezpieczeństwa, skuteczne leki. W swoim rdzeniu, GxP to gwarantuje „leki farmaceutyczne są robione dobrze”. Dobra Praktyka Produkcyjna (GMP) jest najbardziej widoczną częścią GxP w produkcji leków, obejmujące projektowanie sprzętu higienicznego, walidowane procesy, i rygorystyczną dokumentację. Inne komponenty, takie jak GLP, GCP, PKB, i GVP zajmują się różnymi etapami (badania laboratoryjne, próby, dystrybucja, i nadzór nad bezpieczeństwem farmakoterapii, odpowiednio), ale wszystkie mają wspólny cel, jakim jest ochrona pacjentów.
Sprzęt do pakowania farmaceutycznego odgrywa kluczową rolę w przestrzeganiu zasad GxP. Maszyny takie jak napełniacze kapsułek, pakowarki blistrowe, i kartoniarki muszą być zbudowane i sprawdzone pod kątem zgodności ze standardami GMP – na przykład, jest łatwy do czyszczenia, obsługa elektronicznej ewidencji partii, i utrzymanie identyfikowalności. Wybierając Maszyny gotowe do stosowania w GMP i zgodnie z protokołami kwalifikacyjnymi (IR/WH/PQ), firmy mogą zintegrować zasady GxP ze swoimi liniami produkcyjnymi.
Gotowy do zapewnienia zgodności z GxP na Twojej linii produkcyjnej? Weź pod uwagę zaawansowany sprzęt pakujący Jinlu Packing, który jest przeznaczony dla środowisk GMP. Skontaktuj się z Jinlu aby poprosić o wycenę lub dowiedzieć się więcej o naszych maszynach do napełniania kapsułek zgodnych z GMP, pakowarki blistrowe, i linie kartonowe.
Często zadawane pytania dotyczące GxP w branży farmaceutycznej
Co oznacza GxP?
GxP oznacza „Dobrą Praktykę”. Jest to ogólny termin określający różne wytyczne dotyczące jakości w produktach farmaceutycznych. „X” może oznaczać M (Produkcja), L (Laboratorium), C (Kliniczny), D (Dystrybucja), itp. Innymi słowy, Dobra Praktyka Produkcyjna (GMP), Dobra Praktyka Laboratoryjna (GLP), Dobra praktyka kliniczna (GCP), itp..
Czy GMP jest częścią GxP?
Tak. GMP (Dobra Praktyka Produkcyjna) jest jednym z głównych składników GxP. GxP to parasol, a GMP odnosi się konkretnie do produkcji. Kiedy więc mówimy o zgodności z GxP, Często skupiamy się na GMP, ponieważ reguluje ona produkcję i pakowanie. Inne części, takie jak GLP lub GVP, również należą do rodziny GxP.
Jaka jest różnica między GxP i GMP?
GxP to szeroka kategoria wszystkich „Dobrych Praktyk” w farmacji (obejmujące rozwój poprzez dystrybucję). GMP jest tylko jedną z takich praktyk, skupiona na produkcji. Pomyśl o GxP jako o całościowych ramach jakości, oraz GMP jako sekcja zajmująca się standardami hali produkcyjnej.
Kto reguluje zgodność z GxP?
Agencje regulacyjne egzekwują zasadę GxP. W USA, FDA nadzoruje cGMP i GLP, oraz FDA/NIH nadzorują GCP w badaniach klinicznych. W Europie, EMA (oraz organy krajowe, takie jak MHRA) egzekwować wytyczne UE GMP i GCP. WHO publikuje międzynarodowe wytyczne GxP, które przyjmuje wiele krajów. Każdy kraj może mieć swoją własną wersję, ale FDA, EMA, WHO to najważniejsze autorytety, do których odwołuje się świat.
Co jest 21 Część CFR 11 i dlaczego jest to ważne dla GxP?
21 Część CFR 11 to rozporządzenie amerykańskiej Agencji ds. Żywności i Leków dotyczące zapisów elektronicznych i podpisów elektronicznych. Dla GxP, oznacza dowolny skomputeryzowany system generujący zapisy (jak oprogramowanie HMI maszyny lub LIMS) muszą mieć kontrolę, aby zapisy były bezpieczne, oznaczone czasem, i nie da się tego zmanipulować. Na przykład, System sterowania maszyny pakującej w blistry będzie wymagał bezpiecznych loginów i ścieżki audytu zmian parametrów. Zgodność z częścią 11 jest obowiązkowy w USA i obowiązuje w innych regionach (Załącznik UE 11) w systemach elektronicznych.
Czy farmaceutyczne maszyny pakujące muszą być zgodne z GxP?
Absolutnie. Każdy sprzęt używany do produkcji lub pakowania leków musi spełniać kryteria projektowania i walidacji GxP. Mam na myśli maszyny pakujące (Wypełniacze kapsułki, Maszyny pęcherzy, wypełniacze w płynie, kartoniarze, itp.) powinien mieć higieniczną konstrukcję, potwierdzone działanie, i funkcje integralności danych. Na przykład, linia pakująca musi przestrzegać udokumentowanych procedur czyszczenia (wymóg GMP) i korzystaj z systemów rejestrujących dane wsadowe. W wielu przypadkach, dostawcy sprzętu do pakowania reklamują maszyny „zgodne z GMP” lub „gotowe do stosowania przez FDA”.. Przy wyborze sprzętu, producenci wymagają takich elementów, jak części kontaktowe ze stali nierdzewnej, Łatwe czyszczenie, i pełne protokoły IQ/OQ/PQ. Sprzęt Jinlu, Na przykład, został zaprojektowany z myślą o tych standardach, aby wspierać środowisko regulowane przez GxP.
Referencje:
1.Dobre Praktyki Produkcyjne -- KTO
2.TRS 986 – Załącznik 2: Dobre praktyki produkcyjne WHO dotyczące produktów farmaceutycznych: Główne zasady -- KTO
3.Wytyczne WHO dotyczące produkcji farmaceutycznej -- KTO
4.Wytyczne dotyczące dobrej praktyki produkcyjnej i dobrej praktyki dystrybucyjnej: Pytania i odpowiedzi —— Europejska Agencja Leków
5.Wytyczne MHRA dotyczące integralności danych GxP —— gov.uk
6.Przewodnik dobrych praktyk GAMP: Działanie systemów komputerowych GxP —— ispe.org




