製薬業界では, 品質と患者の安全が最優先です. すべてのタブレット, バイアル, またはブリスターパックは、効果を確実にするために厳格な管理の下で製造されなければなりません, 純粋な, そして安全. そこです GxP コンプライアンス 入ってくる. GxP は「」を意味する包括的な用語です。良い x 練習する,」 ここで、「x」は製造業を意味します, 研究室, 臨床, 分布, 等. 本質的には, GxP ガイドラインは規制です 品質システム その製薬会社は 従わなければなりません すべてのプロセスを文書化して管理する. 一緒に, これらの規則により、開発全体を通じて医薬品が一貫して生産され、高品質基準に従って管理されることが保証されます。, 製造, テスト, パッケージング, そして配布.
規制当局のようなもの FDA (私たち. 食品医薬品局), エマ (欧州医薬品庁), 誰が, その他は GxP 標準を施行します. 例えば, FDA の現在の適正製造基準 (CGMP) ルール (21 CFR パート 210 ~ 211) 製造の最低要件を設定する, パッキング, 薬が安全で、正しい成分と強度であることを確認するために薬を保持します。. 同様に, WHO と EU のガイドラインは GMP/GLP/GDP/GVP を世界的にカバーしています. 実際に, 製薬会社が開発する 品質マネジメントシステム および標準操作手順 (SOP) GxP 原則について. 製品のすべてのバッチには、最初から最後まで追跡可能な記録があります. 設備であっても資格が必要です (設置/運用/パフォーマンスの資格 – IQ/OQ/PQ 付き) GxP 要件を満たすことが検証されています.

医薬品における GxP とは何ですか?
頭字語 G×P を表します 良い「x」の練習, どこで 「ぐ」 は「良い」です。 「ぷ」 は「練習」です。 「x」はさまざまな分野を表します. GxP は単一の規制ではなく、品質ガイドラインのグループです. GxP の主要な分野には次のものがあります。:
- 優れた製造業 (GMP): 製品が品質基準に従って一貫して生産および管理されていることを保証します. GMPは製造をカバーします, 装置, 設備, 人員, 汚染を最小限に抑えるための文書化, 取り違え, とエラー.
- 研究室の適正な実践 (GLP): 非臨床臨床検査の実施を管理する (毒物検査のような) データの整合性と再現性を確保するため. GLP は臨床検査が十分に文書化されていることを保証します, 監査可能, そして信頼できる.
- 優れた臨床実践 (GCP): デザインの国際標準, 指揮する, 監視, 臨床試験の報告と. GCP は患者の権利を保護します, データの完全性, 試験結果が信頼できるものであることを保証します.
- 適切な配布慣行 (GDP): ストレージの基準を設定する, 取り扱い, および医薬品の輸送. GDP は、医薬品の品質がサプライチェーン全体で維持されることを保証します。たとえば、, 温度をコントロールすることで, 汚染の回避, バッチの追跡.
- 医薬品安全性監視の適切な実践 (GVP): 製品の市販後に医薬品の安全性を監視し、有害事象を報告するためのガイドラインを提供します。. GVP は製薬会社と規制当局の検出を支援します, 評価, マイナスの副作用を防ぐ.
各 GxP 領域は特定の規制またはガイドラインによって施行されます (例えば。, FDA 21 GMP/GLP/GCP の CFR, GMP/GDP/GVP 用の EU EudraLex, 私はガイドライン, 誰が, 等). 細部は異なりますが, 共通の目標は 医薬品の安全性を確保するために, 効果的, あらゆる段階で高品質を実現.
テーブル: GxP の主な分野(GxP 規制の種類)
| G×Pタイプ |
フルネーム |
製品ライフサイクル段階 |
焦点/目的 |
| GLP |
研究室の適正な実践 |
薬物研究 |
非臨床臨床研究を管理する (毒性試験のような) データの品質とトレーサビリティを確保するため. GLP は検査手順の基準を設定します, データ記録, そして報告. |
| GCP |
研究室の適正な実践 |
臨床試験 |
臨床試験の国際基準 (I-GCP). トライアルデザインをカバー, インフォームドコンセント, 監視, 結果の正確な報告. |
| GMP |
優れた臨床実践 |
製造業 |
医薬品が製造/包装において一貫して製造および管理されていることを保証します. 施設・設備基準を含む, 人事訓練, QC試験. |
| GDP |
適切な配布慣行 |
倉庫保管 & ロジスティクス |
医薬品の適切な保管と輸送を確保する (例えば。, 正しい温度, 安全な取り扱い) サプライチェーンを通じて品質が維持される. |
| GVP |
医薬品安全性監視の適切な実践 |
市販後のモニタリング |
市販薬の継続的な安全性モニタリングに関するガイドライン (有害事象報告, リスク管理, 当局とのコミュニケーション). |
製薬業界で GxP が重要な理由?
GxP コンプライアンスは不可欠です。 命は文字通りそれに依存している. その主な目的は次のとおりです。:
- 患者の安全: GxPに従うことで汚染などのリスクを最小限に抑えます, 取り違え, または投与ミス. 例えば, GMPルール (適切な衛生管理やプロセス管理など) 製品間の相互汚染の防止に役立ちます. GxPなし, 標準以下の医薬品は患者に害を及ぼすか、効果が不十分になる可能性があります. 規制当局は次のことを強調しています 「GxP ルールが存在する理由は 1 つあります」: 患者を守るために」.
- 製品の品質と一貫性: GxP フレームワークにより、医薬品のすべてのバッチが同じ仕様を満たしていることが保証されます. これには一貫した効力が含まれます, 純度, および安定性. 厳格な管理を徹底することで、 (例えば. 機器の校正, 検証されたメソッド, 工程内テスト), メーカーは、各バイアルまたは錠剤がそのラベルの主張と一致することを保証できます。.
- 規制遵守と信頼: GxP の遵守は法的に義務付けられています. FDAなどの機関, エマ, MHRA, 総合機構, と WHO は GxP の検査と監査に依存しています. 強力な GxP 実践を実証する企業は規制当局からの信頼を築きます. 違反すると警告書が発行される可能性があります, 罰金, 製品リコール, あるいはさらに悪いことに. ある業界ガイドによると, 規制当局は、製品の完全性と患者の安全性を強調する「GxP 規格への準拠を検証」するために監査を実施します。.
- データの整合性: GxP の重要な側面は、データが正確で信頼できるものであることを保証することです. 現代の規制 (例えば. FDAの 21 CFRパート 11, EU附属書 11) 電子記録の安全性を要求する, タイムスタンプ付き, そして改ざん防止. 重要な原則のようなもの アルコアプラス (起因する, 読みやすい, 同時代の, オリジナル, 正確な, プラス完了, 一貫性のある, 永続的な, 利用可能) データの品質を判断するために使用されます. 実際に, これはソフトウェアに監査証跡を実装することを意味します, マスタードキュメントのロック, 定期的にバッチ記録を確認します. 堅牢なデータ整合性は GxP の基礎です; それなしで, たとえ良く作られた製品であっても信頼されない.
要するに, GxP は医薬品の品質管理の基盤です. それは次のことを保証します あらゆるステップ — ラボでのテストから最終包装まで — 管理された下で行われます, 文書化された条件. 例えば, あるガイドは次のように観察しています。 「GxP コンプライアンスにより、医薬品と生物製剤の開発が保証されます」, 製造された, 厳格な基準に従ってテストされています,」 コストのかかる汚染やラベル貼り間違いの防止. したがって, GxP は患者を保護するだけでなく、製薬会社の信頼できるブランド評判と市場アクセスを支えます。.

GMP について – 最も重要な GxP 規格
すべての GxP 領域が重要である一方で、, GMP (優れた製造業) 多くの場合、医薬品製造コンプライアンスの基盤とみなされます. GMPは医薬品の製造プロセス全体を対象としています, 原材料も含めて, 装置, 設備, プロセス, および包装ライン. その中心的な目標は、 リスクを最小限に抑える 厳格な管理と文書化を実施することにより、製品の汚染または逸脱を防止します. GMP の主要な要素には次のものがあります。:
- 施設・設備の設計: 機器は衛生的に操作できるように設計する必要があります. これは製薬グレードの材料を使用することを意味します (例えば. 316Lステンレス), 滑らかな溶接, デッドゾーンなし, 掃除に簡単にアクセスできます. 包装機用, これには片持ちコンベヤーが必要になる場合があります, クイックリリースパーツ, そして囲まれた警備員. 適切なレイアウトにより混同が防止され、洗浄の検証が容易になります (検証された洗浄手順).
- ユーティリティとキャリブレーション: すべてのユーティリティ (水, 圧縮空気, 電気) 品質仕様を満たす必要がある. 機器とセンサー (天秤, 流量計, 温度プローブ) 定期的に必要です 較正 書面による手続きの下で. FDA は重要な機器の校正記録を明示的に期待しています. 例えば, 充填ボトルのラインでは、各用量が正確であることを保証するために容積ポンプが校正されます。.
- 検証と認定: GMP は、機器とプロセスが認定され、検証されることを義務付けています. これには設置資格が必要です (IQ), 運用資格 (OQ), およびパフォーマンス資格 (PQ) システムが意図したとおりに動作することを証明するため (以下で詳しく説明します). 例えば, 新しいブリスター包装機はシール温度を確認するために IQ/OQ/PQ テストを受ける必要があります, シールの完全性, インデックスシステムは仕様を満たしています. すべての重要なマシン (カプセル充填剤, 箱詰め業者, 液体充填ライン) この検証ライフサイクルに従う必要があります.
- 標準操作手順 (SOP): 詳細, 製造ステップには書面によるSOPが必須です, クリーニング, メンテナンス, と品質管理. オペレーターはこれらの SOP に基づいてトレーニングを受ける必要があり、いかなる変更も正式な変更管理システムを通じて管理する必要があります。. 監査人は最新の SOP とトレーニング記録を探します.
- 適切な文書化の実践: GMPは次のことを強調しています 「文書化されていない場合, それは起こらなかった。」 全ての手続き, バッチレコード, ログのクリーニング, そしてQCテストは読みやすく同時に記録されなければなりません. 完成したバッチ製造記録 (BMR) 材料の調整を含める必要があります, 機器の設定, 工程内チェック, および逸脱/修正. 現代のトレンドはバッチ記録の電子化を推進 (eBR) 監査証跡あり 21 CFRパート 11.
- 品質管理 (品質管理) およびバッチリリース: QC試験 (例えば. 効力, 無菌性, 識別) 原材料と完成品に対して行われます. QA/QC 部門がすべての文書をレビューし、テスト結果を確認した後でのみ、バッチがリリースされます。. この最終チェックはGMPの重要な部分です.
GMP包装に関するJinlu Packingのブログ これらの要件のいくつかを強調表示します. 例えば, それはそれを指摘します 予防保守と校正 GMPによって義務付けられています (21 CFR 211.68 定義された間隔と記録が必要です). それはまた強調します オートメーション & エラー防止: 最新のGMP包装ラインは視覚検査を使用しています, バーコードスキャン, ヒューマンエラーを回避するためのインターロック. データの整合性とコンピューター制御も GMP の対象となります: ライン上のすべてのコンピュータ化されたシステム (例えば. PLC, ビジョンシステム, HMIS) に従わなければなりません 21 CFRパート 11 – 一意のログインを意味します, 監査証跡, 電子署名, 安全な記録保持. 実際に, これは、マシンの制御ソフトウェアがすべてのパラメータ変更をタイムスタンプとユーザー ID とともに記録することを意味します。, バッチを承認するにはマネージャーの電子署名が必要です.
包装機器の GMP チェックリスト
GMP要件をチェックリスト形式で表示すると便利です. 包装機械用, メーカーは通常、:
- デザイン & 材料: 衛生的な構造 (ステンレス鋼部品, FDA承認のシール), 「死んだ」領域がない, 傾斜面, 掃除のために簡単に分解できる.
- 検証: 各マシンで IQ/OQ/PQ を完了する. 文書化された機能のテスト (封印, 充填, 重さを量る) そしてパフォーマンス. (以下の「機器の認定」セクションを参照してください。)
- クリーニング: 綿棒またはすすぎテストによる検証済みの洗浄手順 (通常は TOC または特定のアッセイを使用します) そして記録.
- メンテナンス & 較正: ログ付きの予防保守スケジュール. 定量ポンプの校正, 天秤, 精度を確保するためのセンサー.
- コントロール & オートメーション: 画像検査システム (例えば。, 金属探知機, 重量チェッカー) 欠陥を検出する, ミスフィードを防ぐセンサー, ドアが開くと停止するインターロック.
- データの整合性: 21 CFRパート 11 コンプライアンス – 監査証跡, ユーザーアクセス制御, 電子署名, 安全なデータ. すべての記録 (SOP, バッチデータ, 逸脱) 変更不可能に保存される.
- トレーサビリティ: シリアル化/UIDのサポート, バーコーディング, 各プライマリ パックをバッチ レコードにリンクする, 必要に応じてリコールを有効にする. (Jinlu マシンには、トレーサビリティのためにラベル プリンタやコード リーダーが組み込まれていることがよくあります。)
- 環境 & ラインコントロール: 必要に応じて適切なクリーンルーム基準, バッチ間のラインクリアランスチェックを文書化, 取り違えを防ぐための適切なラベルと材料の取り扱い.
これらのコントロールに従うことで, メーカーは自社の包装機器がGMPガイドライン内で動作することを確信できます。. 例えば, カプセル充填機 そして ブリスター包装機 Jinlu の製品は GMP 対応機能を備えて構築されています (滑らかなGMP表面, CIP機能, 等) これらの要求を満たすために.

製薬機器メーカーの GxP 要件
包装機器サプライヤーは GxP において重要な役割を果たします. 購入者は機械が堅牢で効率的であるだけではないことを期待しています, 規制遵守を容易にするためにも構築されています. 主な要件には以下が含まれます:
- 資格取得サポート (IR/WH/PQ): ベンダーは提供する必要があります プロトコル そして資格取得の補助も. これは、文書化された設置資格を意味します。 (IQ) マシンが正しくインストールされていることを示す, 運用資格 (OQ) 仕様通りに機能することを証明するため, およびパフォーマンス資格 (PQ) 一貫して許容可能な出力が生成されることを確認する. 例えば, Jinlu Packing は、IQ/OQ/PQ キットと完全な FAT/SAT プロトコルを備えた各マシンを提供します。. これらのテンプレートは、多くの場合、購入者の QA チームによって調整できます。, 検証中の時間を節約する. ベンダーは認定試験に参加したり、認定機器チェックリストを提供したりする場合もあります。.
- ドキュメントパッケージ: 機械と一緒に, サプライヤーは完全な書類を提出する必要があります. 代表的な項目としては、 ユーザーマニュアル, マスターパーツリスト, 電気回路図, そして メンテナンス手順. 重要なこと, 脂肪 (工場受け入れテスト) そして 土 (サイト受け入れテスト) レポートは、機械が工場および現場のテストに合格したことを文書化します。. あらゆる測定装置の校正証明書を含める必要があります. 実際に, 準拠したドキュメント パッケージにはシステム仕様も記載されています, クリーニングSOP, リスク評価, および変更管理履歴.
- データ整合性機能: 最新の機器は、GxP データ規格に準拠した電子制御を提供する必要があります. これには安全なユーザー アカウントが含まれます (ロールベースのアクセス), 重要なアクションに対する必須の電子署名, パラメータへの変更の完全な監査証跡. 例えば, HMI 画面では、生産を開始するためにシフト監督者のログインが必要になる場合があります, すべてのレシピや設定変更にはタイムスタンプが付けられます. 機械はデジタルバッチレコード出力もサポートする場合があります, MES/ERPシステムとの統合.
- 検証およびテストツール: 一部のベンダーにはデータログ用のソフトウェアツールが含まれています, 較正, または検証. これは、センサーテストを実行するためのプリインストールされたソフトウェアである可能性があります, または、PQ 実行中にパラメータをロックする組み込み機能. これらの機能により、検証中の手動作業が軽減されます。.
- 衛生的で安全な設計: 機器は掃除とメンテナンスが簡単でなければなりません. クイックリリースパーツなどの機能, 無製品ゾーン, およびCIP (定置洗浄) オプションは洗浄の検証を満たすのに役立ちます. 製品と接触する材料は不活性でなければなりません (例えば. 316LSS, FDA認可のプラスチック). 安全ガードとインターロックがオペレーターを保護, コンプライアンスも確保します (例えば. 開けると機械が止まる).
- アフターセールスサポート: GxP コンプライアンスは継続中です. メーカーは定期的な再認定または再校正を必要とする場合があります. サプライヤーはライフサイクルサービスを提供する必要があります: スペアパーツ (検証済みの部品を迅速に交換するため), 保守契約, 変更が発生した場合は検証ドキュメントも更新されます. オンサイトで認定サービスを提供するサプライヤーの意欲 (IR/WH/PQ) コンプライアンスの取り組みを大幅に円滑化できる.
機器の適格性チェックリスト: 以下の表は、新しい製薬機械を認定するための一般的な手順と文書をまとめたものです。:
| 段階 |
主な活動 |
代表的な書類 |
| ユーザー要件 (URS) |
重要な仕様を定義する (例えば. 出力レート, 正確さ) |
ユーザー要求仕様 |
| 設計適格性評価 (DQ) |
ベンダーの設計が URS を満たしていることを確認する |
設計仕様レビューレポート |
| 工場での受け入れ (脂肪) |
主要機能の工場テスト, 多くの場合、IQ/OQ テストが反映されます |
FATレポート |
| 設置資格 (IQ) |
正しいインストールを確認する: 公共事業, 機械的なセットアップ, ドキュメント (図面, 証明書) |
IQプロトコル & チェックリスト |
| 運用資格 (OQ) |
すべての機能をテストする: 空ラン性能, コントロール, アラーム, 安全機能 |
OQプロトコル & 結果 |
| パフォーマンス資格 (PQ) |
実際の製品を使用して完全な運用を実行する: 出力品質をチェックする, 一貫性, ストレス状態 |
PQプロトコル, 走行記録, サンプルテスト結果 |
| 最終リリース |
すべての資格記録を確認する; 機械をGMP生産に導入するためのQA承認 |
資格概要レポート |
(注記: Jinlu のマシンには以下が付属します IQ/OQ/PQ テンプレート そして完全な 工場での受け入れ ドキュメントパッケージ, 顧客は必要に応じて適応できます。) ユーザー要件から PQ に至るこの構造化されたアプローチは、次のような規制によって義務付けられています。 EU GMP 付属書 15 およびFDAガイドライン. いずれかのステップをスキップすると、コンプライアンス ギャップが生じる可能性があります.

GxP コンプライアンスの一般的な課題
明確なルールがあっても, 企業は GxP コンプライアンスのハードルに直面することがよくあります. よくある課題には次のようなものがあります。:
- 不完全な検証または文書化: 最も深刻なギャップの 1 つは、完全な IQ/OQ/PQ 記録のない機器を使用していることです。. 「検証をスキップする」,」または持っている 部分的 資格, とみなされます 「致命的なGMPギャップ」. 同様に, 欠落またはずさんなバッチ記録と SOP はコンプライアンスを損なう: 検査官は文書を探すように訓練されています. ある専門家が言うように: 「書かれてなかったら, それは起こらなかった。」 記録管理が不十分 (失われたファイル, 判読不能なメモ, 古いバージョン) それは一般的な危険信号です.
- 機器のメンテナンスと校正の失効: 規制当局は校正期限の期限切れや修理の延期などの問題を繰り返し発見. 壊れたセンサーや校正されていないスケールは、疑わしいデータや製品につながる可能性があります. (例えば, ソーコル氏の分析によると、忘れられた校正や磨耗した部品は、 「単純な失敗」 バッチホールドがトリガーされる可能性があります。) 保守ログの徹底, 校正スケジュールにデジタル トラッカーを使用する, この問題を克服するには、スタッフに問題をすぐに報告できるようにすることがベスト プラクティスです。.
- データの整合性の問題: 最新の GMP ラインは電子システムに依存しています. データ管理の失敗は GxP を破壊する可能性があります. 例としては、無効化された監査証跡が挙げられます。, 弱いパスワード, 元のエントリの代わりにデータをコピー/ペーストしました, または電子ログの確認を怠った場合. 企業は ALCOA+ 原則を強制する必要があります。, すべてのデータ入力が正しいことを確認します 起因する (ユーザーにリンクされている), 読みやすい (クリア), 同時代の (リアルタイムで記録される), オリジナル/正確, そして 完全/一貫性のある. オペレーターのトレーニングと可能な場合の自動化 (例えば. コンピュータでロックされた記録) 手動による上書きや省略を防ぐのに役立ちます.
- 変更管理と CAPA の不備: あらゆる変更には堅牢な変更管理プロセスが必要です (装備のアップグレード, 新しいSOP, 新しい原料). よくある間違いは、変更の文書化を怠ったり、変更後の再検証をスキップしたりすることです。. 同様に, 逸脱を適切に調査できなかった (根本原因の分析を行わずに問題を単なる「人為的エラー」として片付ける) 問題が長引く可能性がある. 規制当局は強力な CAPA を期待している (是正措置と予防措置) あらゆる逸脱に対処するシステム.
- トレーニングと文化の問題: GxP では、すべての担当者が訓練を受け、品質の高い手順を認識していることが求められます. 不適切な研修プログラムや離職率の高さは、意図しない違反につながる可能性があります. 品質文化の構築 (スタッフがコンプライアンスに対する責任を感じ、問題を報告することが奨励される場所) 不可欠だが開発が遅いことが多い.
要約すれば, 課題は組織的なものであることが多い: ドキュメント, メンテナンス, トレーニング, そしてデータプラクティス. それらを克服するにはシステムへの投資が必要です (電子文書管理ソフトウェアや校正追跡ソフトウェアなど), 規律あるSOP, 頻繁な内部監査. これらの分野に積極的に取り組む企業は、公的検査の際に GxP コンプライアンスをよりスムーズに行うことができます。.
製薬会社が GxP コンプライアンスを達成する方法
GxP コンプライアンスの達成は、 組織全体にわたるプロジェクト. 以下は一般的な手順のシーケンスです (フローチャートで示される) 製薬会社が準拠したシステムを構築するために従うこと:
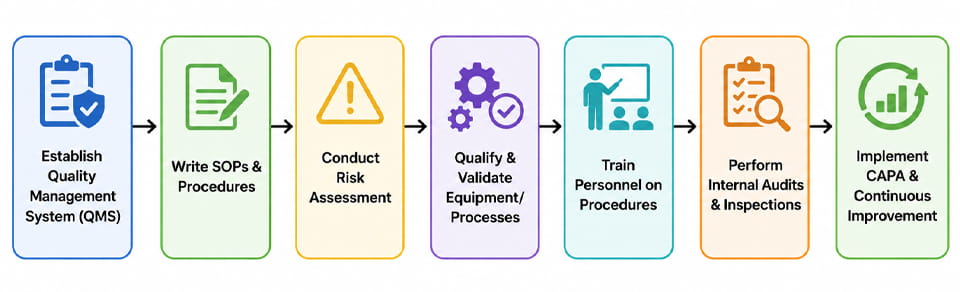
- 品質マネジメントシステム (QMS): 品質のための組織構造を定義することから始めます (例えば. 品質マニュアル, 政策). これには、質の高い役割と責任の割り当てが含まれます.
- SOPの開発 & ドキュメント: 生産のための標準手順の草案と承認, テスト, 変更制御, 逸脱, 等. 各プロセスを明確に文書化する.
- リスクアセスメント: 正式なリスク評価を実行する (例えば. FMEA) 重要なプロセスパラメータと制御を特定するため. これにより、どこに焦点を当てて検証とモニタリングを行うべきかがわかります。.
- 装置/プロセスの認定 (IR/WH/PQ): 前のセクションと同様に, すべての製造装置とプロセスを認定する. 詳細な検証プロトコルとレポートを維持する.
- トレーニング: 電車の運転士, エンジニア, 承認された手順に関する QA/QC スタッフ, GxP 原則, そして設備の使用.
- 内部監査: 定期的な自己検査または模擬監査を実施して、遵守状況を確認し、問題を特定します。 (例えば. バッチレコードをチェックする, 環境ログ, 校正ステータス).
- キャパ: 逸脱または所見が特定されたときは常に (内部または規制当局によって), 根本原因を調査する, 是正措置を適用する, 再発防止のため手順を更新する.
- 継続的な改善: データの使用 (例えば. 製造報告書やクレームログからの傾向分析) 品質向上を推進し、プロセスを最適化する.
各ステップは必要に応じてループバックします。, 監査中に変更が見つかった場合は、SOP の更新と再トレーニングが必要になります. 企業も品質リスク管理を活用しています (QRM) デザインによる品質 (QbD) このアプローチを体系化するための原則. の 上のフローチャート この循環プロセスを示しています.
GxPと包装機器の関係
モダンな 医薬品 パッケージライン 複雑であり、GxP 原則を直接具体化する必要がある. ボトルスクランブラーからブリスターマシン、カートナーに至るまで、ライン上のすべての機械は、GMP を遵守する方法で設計および使用されなければなりません。. 主要な接続は次のとおりです:
- 衛生設計: 包装機は汚染を避けるように作られています. 例えば, ある ブリスター包装機 密閉された成形セクションと滑らかなフィードトラックを備えています, 製品が床や埃っぽい表面に接触しないようにする. 製品接触領域のツールと部品は通常、ステンレス鋼または医療グレードのプラスチックです。, GMP 材料要件との整合.
- 検証対応: 包装機器は完全に認定可能である必要があります. サプライヤーは検証を容易にするために機械を設計することがよくあります。: センサーがアクセスできるエリア (校正チェック用), 空の実行と完全な実行を実行する機能, そして安定したパフォーマンス. 顧客は機械に仕様があることを期待しています (SOPとマニュアル) IQ/OQ/PQ 活動に直接結びつく.
- バッチ記録とトレーサビリティ: GMP ラインのすべてのステップが文書化されます. 自動包装機は多くの場合、バッチ番号を記録するソフトウェアと統合されています。, 回線速度, そして出力. 例えば, 瓶詰めラインでは、各ボトルにバッチ コードとタイムスタンプが自動的にラベル付けされる場合があります。. これらのコードは実稼働環境にリンクされます。. システムは重量や不合格品の数を記録することもできます (例えば. インライン重量チェッカーによって充填不足のカプセルにフラグが付けられる). このデータは電子バッチ記録の一部になります. 要するに, 包装機はメンテナンスに役立ちます トレーサビリティ すべてのユニットの, これは規制要件です.
- 21 CFRパート 11 コンプライアンス: 前述したように, 包装機器のコンピューター制御 (ヒューマンマシンインターフェースやPLCなど) 電子記録規則を遵守する必要があります. これは、オペレーターが一意の ID を使用してログインすることを意味します。, 許可なくパラメータを変更することはできません. データログ (例えば. 設定, テスト結果) 安全でタイムスタンプが付いている必要がある. 現在、多くの最新マシンにはユーザー アクセス レベルが含まれています (演算子 vs. 監督者) これらのニーズを満たす監査ログ機能.
- エラーの削減と自動化: 自動梱包により手作業が軽減されます, ヒューマンエラーを減らす (GxP に焦点を当てる). 例えば, カプセル充填機 Jinlu の製品は正確な投与で高速に実行できます, 手動修正の必要性を最小限に抑える. 同じく, 自動箱詰め機 一貫したシールを確保する. コンプライアンスのために, これは、コンポーネントの選択を間違えたり、ラベルを間違えたりする可能性が少なくなることを意味します。これは、異なる製品を並べて梱包する場合に重要です。.
- 規制上の特徴: DSCSA などの新しい規制 (アメリカ合衆国) または口蹄疫 (欧州連合) 個々のパックのシリアル番号を要求する. 包装機には多くの場合、これらのコードを適用/検証するために 2D コード プリンターとビジョン カメラが装備されています。. このような特徴は、市場の法則を示しています。 (GxP トレーサビリティの名の下に) 形状機器の機能.
- コンプライアンスサポート: ジンルの装備, 例えば, GMP 対応であり、多くの場合、認定とトレーサビリティのサポートが付属しています. 一般的なカプセル充填剤には CIP が搭載されている場合があります。 (定位置クリーン) システムと滅菌用の取り外し可能な飼料ホッパー. a 水疱ライン 開いたときに作動を防ぐ安全インターロック付きのガードドアが含まれる場合があります. これらの設計詳細は GMP を直接サポートします.
GxPを考慮した包装機械の選択, 製薬会社がコンプライアンスをスムーズに. 例えば, Jinlu のインストール カプセル充填機 または 箱詰め機 購入者が製薬基準に基づいて構築された機械をすでに所有していることを意味します, ドキュメント付き (FAT/SATのような) 検証の準備ができています. 最終的に, 適切に設計された装置は、準拠した製造プロセスの要です.

結論
GxP は医薬品の品質の基盤です. 単なるルールではありません, しかし、安全性を確保するための全社的な取り組み, 効果的な薬. その核心, GxP は次のことを保証します。 「医薬品は正しく作られています。」 優れた製造業 (GMP) 医薬品生産のための GxP の最も重要な部分です, 衛生機器の設計をカバー, 検証済みのプロセス, そして厳格な文書化. GLP などのその他のコンポーネント, GCP, GDP, と GVP はさまざまな段階に対応します (研究室での研究, 試練, 分布, および医薬品安全性監視, それぞれ), しかし、患者を守るという目標は全員が共有しています.
医薬品包装設備 GxP コンプライアンスにおいて重要な役割を果たします. カプセル充填機などの機械, ブリスターパッカー, また、カートナーは GMP 基準を満たすように製造および検証される必要があります。たとえば、, 掃除が簡単であること, 電子バッチ記録のサポート, トレーサビリティの維持. 選択することで GMP対応機械 および以下の認定プロトコル (IR/WH/PQ), 企業は GxP 原則を自社の生産ラインに統合できます.
生産ラインで GxP コンプライアンスを確保する準備ができています? Jinlu Packing の高度な包装設備を検討してください, GMP環境向けに設計されています. ジンルに連絡する 見積もりをリクエストしたり、GMP 準拠のカプセル充填機について詳しく知りたい場合は, ブリスターパッカー, および箱詰めライン.
GxP in Pharma に関するよくある質問
GxP とは何の略ですか?
GxP は「グッド プラクティス」の略です. 医薬品におけるさまざまな品質ガイドラインの総称です。. 「x」には M を指定できます (製造業), l (研究室), C (臨床), D (分布), 等. 言い換えると, 優れた製造業 (GMP), 研究室の適正な実践 (GLP), 優れた臨床実践 (GCP), 等.
GMP は GxP の一部ですか?
はい. GMP (優れた製造業) GxP の主要コンポーネントの 1 つです. GxPは傘です, GMP は特に製造を指します。. したがって、GxP コンプライアンスについて話すときは、, GMP は生産と包装を管理するため、多くの場合最大の焦点となります。. GLP や GVP などの他の部品も GxP ファミリに含まれています.
GxP と GMP の違いは何ですか?
GxP は製薬業界におけるすべての「グッドプラクティス」の広範なカテゴリーです (開発から流通までをカバー). GMP はその実践の 1 つにすぎません, 生産に焦点を当てた. GxP を品質フレームワーク全体として考える, 製造現場の基準を扱う部門としてのGMP.
GxP コンプライアンスを規制するのは誰ですか?
規制当局は GxP を施行します. アメリカでは, FDA は cGMP と GLP を監督します, FDA/NIH は臨床試験における GCP を監督します. ヨーロッパでは, エマ (および MHRA のような国家機関) EU GMP および GCP ガイドラインを施行する. WHO、多くの国が採用する国際的な GxP ガイダンスを公開. 各国には独自のバージョンがある場合があります, しかしFDA, エマ, WHOは世界的に参照される主要な当局です.
何ですか 21 CFRパート 11 そしてなぜそれが GxP にとって重要なのか?
21 CFRパート 11 電子記録と電子署名に関する米国 FDA の規制です。. GxP向け, 記録を生成するコンピュータ化されたシステムを意味します (機械の HMI または LIMS ソフトウェアのような) 記録を安全に保つためには制御が必要です, タイムスタンプ付き, そして改ざんすることはできません. 例えば, ブリスター包装機の制御システムには安全なログインとパラメータ変更の監査証跡が必要です. 部品の適合性 11 米国では必須であり、他の地域でも指導されています (EU附属書 11) 電子システムについて.
医薬品包装機は GxP に準拠する必要がありますか??
絶対に. 医薬品の製造または包装に使用される機器はすべて、GxP の設計および検証基準を満たしている必要があります. 包装機械のことです (カプセル充填剤, ブリスターマシン, 液体充填剤, 箱詰め業者, 等) 衛生的なデザインである必要があります, 検証済みの操作, データ整合性機能. 例えば, 包装ラインは文書化された洗浄手順に従わなければなりません (GMP要件) バッチデータを記録するシステムを使用する. 多くの場合, 包装機器ベンダーは「GMP 準拠」または「FDA 対応」の機械を宣伝しています. 装備品を選ぶときは, メーカーはステンレス鋼の接触部品などの機能を要求します, 簡単な掃除, および完全な IQ/OQ/PQ プロトコル. ジンルの装備, 例えば, GxP で規制された環境をサポートするために、これらの標準を念頭に置いて設計されています。.
参考文献:
1.適正製造基準 - 誰が
2.TRS 986 – 別館 2: WHO の医薬品の適正製造基準: 主な原則 - 誰が
3.医薬品製造に関するWHOのガイドライン - 誰が
4.適正製造慣行および適正流通慣行に関するガイダンス: 質疑応答 —— 欧州医薬品庁
5.GxP データの整合性に関する MHRA ガイダンス —— 英国政府
6.GAMP グッドプラクティスガイド: GxPコンピュータ化システムの運用 —— ispe.org




