Was ist Verpackungsvalidierung in der Pharmaindustrie?? Ein vollständiger Leitfaden für Pharmahersteller
Was ist Verpackungsvalidierung in der Pharmaindustrie?? Ein vollständiger Leitfaden für Pharmahersteller
April 29, 2026
Keine Kommentare
In der Pharmaindustrie, Verpackungsvalidierung ist eine kritische Teilmenge von Prozessvalidierung Der Schwerpunkt liegt darauf, sicherzustellen, dass Verpackungsanlagen und -prozesse die Qualität von Arzneimittelprodukten dauerhaft schützen. Dabei handelt es sich um ein dokumentiertes Qualifizierungsprogramm (Installation, Betriebsbereit, Leistung) und testen (z.B. Integrität des Behälterverschlusses, Stabilität, Transport) um nachzuweisen, dass Verpackungssysteme unter GMP-Bedingungen wie vorgesehen funktionieren. Eine wirksame Verpackungsvalidierung gewährleistet die Patientensicherheit (durch Verhinderung von Kontaminationen, Beeinträchtigungen oder Verwechslungen) und stellt die Einhaltung gesetzlicher Vorschriften sicher (FDA, EMA, WER, ICH, ISO). Dieser Leitfaden erklärt die Definition, Schritte, Tests, Standards, Und Best Practices zur Validierung von Pharmaverpackungen, und zeigt, wie automatisierte Geräte (wie Maschinen von Jinlu Packing) ist darauf ausgelegt, diese strengen Anforderungen zu erfüllen.
Definition der Verpackungsvalidierung
Verpackungsvalidierung im Pharmabereich mit dokumentierten Beweisen demonstrieren dass der Verpackungsprozess (Ausrüstung und Materialien) stellt zuverlässig eine Verpackung her, die das Arzneimittel schützt. In der Praxis, Es werden dieselben strengen Validierungskonzepte angewendet, die auch in der Fertigung zum Einsatz kommen (IR/WH/PQ) Zu primär, sekundär, und Tertiärverpackung Operationen. Gemäß den GMP-Leitlinien der WHO, „Verpackungsprozesse und -geräte müssen auf die gleiche Weise validiert/qualifiziert werden wie jeder andere Teil der Verarbeitung in einer pharmazeutischen Anlage.“. Mit anderen Worten, Die Verpackungsvalidierung stellt sicher, dass Behälterverschlusssystem, Beschriftung, und Verpackungslinien Qualitätsvorgaben und behördliche Anforderungen konsequent erfüllen. Das Ziel besteht darin, die Wirksamkeit des Medikaments aufrechtzuerhalten, Reinheit und Stabilität durch Verpackung, Verteilung und Haltbarkeit, und verhindert gleichzeitig Verwechslungen, Fälschungen, oder Beschädigung.
Zu den typischen Leistungen der Verpackungsvalidierung gehören: Spezifikation der Benutzeranforderungen (URS) (das Design & Qualitätskriterien), Installations-/Betriebs-/Leistungsqualifizierungsprotokolle, und Prüfprotokolle. Der Validierungsprozess umfasst:
Primärverpackung (Der Behälter oder die Barriere kommt direkt mit dem Medikament in Kontakt, z.B. Fläschchen, Blasenhöhlen, Beutel)
Sekundärverpackung (z.B. Kartons, Etiketten, Einsätze, Serialisierung, die primäre Pakete gruppiert)
Tertiärverpackung (Großtransportbehälter, Paletten, Kühlkettenverpackung für den Transport)
Jede Ebene muss das Produkt schützen. Zum Beispiel, die australische TGA definiert „Primärverpackung“ (der Behälter, der die Ware unmittelbar abdeckt) und unterscheidet Sekundärverpackung als Umkarton oder Umhüllung. Letztlich, Die Verpackungsvalidierung knüpft an GMP an – und stellt dies sicher Behälter und Verschlüsse verändern das Arzneimittel nicht Und „angemessenen Schutz vor äußeren Einflüssen bieten“ während der Lagerung und Verwendung.
Warum die Verpackungsvalidierung in der Pharmaindustrie von entscheidender Bedeutung ist
Pharmazeutische Verpackungen sind der letzte Schutz für die Arzneimittelqualität. Die Validierung von Verpackungsprozessen ist aus diesem Grund von entscheidender Bedeutung:
Gewährleistet Produktsicherheit und -integrität: Eine ordnungsgemäß validierte Verpackung verhindert eine Kontamination, Eindringen von Feuchtigkeit, oder Sauerstoffeinwirkung, die Wirkstoffe abbauen könnte. Zum Beispiel, Durch Lecks oder Dichtungsfehler könnten Mikroben oder Gase eindringen, Unfruchtbarkeit oder Potenz beeinträchtigen.
Vorschriftenregulierung: Agenturen (FDA, EMA/ICH, WER) erfordern eine validierte Verpackung. UNS. FDAs 21 CFR 211.94 verbietet Schließungen, die die Arzneimittelqualität beeinträchtigen, und schreibt Schließungen vor „angemessenen Schutz vor vorhersehbaren äußeren Faktoren bieten“. EU-GMP-Anhang 1 beharrt ebenfalls darauf „Behälter sollten mit entsprechend validierten Methoden verschlossen werden“ (mit 100% Integritätsprüfung für fusionsgeschlossene Produkte). Die WHO weist ausdrücklich darauf hin, dass Verpackungssysteme wie alle anderen GMP-Prozesse qualifiziert sein müssen. Nichteinhaltung kann bedeuten erinnert sich, Importablehnungen oder Prüfungszitate.
Verlängert die Haltbarkeit: Unter Stabilitätsbedingungen validierte Verpackungsmaterialien und Siegel helfen bei der Festlegung genauer Angaben zur Haltbarkeit. Die ICH-Q1A-Leitlinie legt sogar fest, dass Stabilitätsstudien in der EU durchgeführt werden müssen „Behälterverschlusssystem“ für das Produkt vorgeschlagen. Wenn die Verpackung versagt, Stabilitätsdaten sind ungültig.
Serialisierung und Fälschungsschutz: Moderne Pharmaverpackungen beinhalten häufig eine Serialisierung, manipulationssichere Siegel, und Fälschungsschutzfunktionen. Validierung dieser Funktionen (z.B. Scanbarkeit von Barcodes, Integrität von Manipulationsbändern) trägt zur Sicherung der Lieferkette bei. Ein Fehler bei der Etikettierung oder Serialisierung kann die Produktfreigabe blockieren.
Qualität und Kosteneffizienz: Effiziente validierte Linien reduzieren Fehler (Fehlfüllungen, Etikettenfehler, zerdrückte Kartons) und Schrott. Automatisierte Inspektionen (Bildverarbeitungssysteme zur Siegel-/Etikettenprüfung) sind oft Teil validierter Kontrollen. Gesamt, Validierte Verpackungsprozesse minimieren das Risiko für den Patienten und stellen eine konsistente Versorgung mit wirksamen Produkten sicher, authentische Medikamente.
Schlüsselfaktoren für die Validierung
Patientensicherheit: Eine unsachgemäße Verpackung kann Patienten schaden (z.B. kontaminierte Injektionspräparate). Die Validierung dokumentiert die Sicherheitsbarriere.
Regulierungsrisikominderung: Bei nicht validierten Prozessen besteht ein hohes Risiko für Rückrufe oder Sanktionen. Regulierungsbehörden erwarten Qualifikationsdaten für jede Produktionslinie.
Geschäftskontinuität: Verpackungsfehler bei der Verteilung von Abfallprodukten und -zeit. Validierung stellt sicher „richtig gleich beim ersten Mal“ Betrieb unter GMP.
Audit-Bereitschaft: Bei Audits durch Qualitätsbehörden oder Kundenaufsichtsbehörden werden im Rahmen von GMP-Inspektionen Verpackungsvalidierungsaufzeichnungen und Gerätequalifikationen überprüft.
Umfang der Validierung pharmazeutischer Verpackungen
Die Verpackungsvalidierung umfasst alle Verpackungsstufen. Typischerweise, wir unterscheiden:
Validierung der Primärverpackung: Konzentriert sich auf den Behälterverschluss (Fläschchen, Ampullen, Blasenhöhlen, Flaschen). Zu den Tests gehört auch die Integrität der Versiegelung, Materialkompatibilität (extrahierbare/auslaugbare Stoffe) und Leistung des Behälterverschlusses. Für sterile Produkte, Integritätstests für Behälterverschlüsse (CCIT) sind zwingend erforderlich, um die Aufrechterhaltung der Sterilität sicherzustellen.
Validierung der Sekundärverpackung: Deckt die unmittelbare Außenverpackung ab (Kartons, Etiketten, Flugblätter, Trockenmittel). Dazu gehört auch die Prüfung der Lesbarkeit/Genauigkeit des Etiketts, Falltests für Kartons, und Boxkompressionstests (für den Transport). Durch die Etikettenüberprüfung wird sichergestellt, dass die richtigen Patienteninformationen konsistent angewendet werden.
Tertiäre Verpackungsvalidierung: Beinhaltet Groß-/Transportverpackung. Hier, Leistungstests wie Vibration, fallen, und Temperaturwechsel (Kühlkette) Simulieren Sie Transportbelastungen, um zu überprüfen, ob Paletten und Versandkartons während des Versands die Produktintegrität und -etikettierung bewahren. Standardprotokolle wie ASTM D4169 (Transportsimulation) werden häufig verwendet.
Jede Stufe erfordert eine Risikobewertung und entsprechende Tests: Zum Beispiel, primär Die Verpackung könnte ein Alleinstellungsmerkmal sein <1207>-Stilintegritätstests, während sekundär Bei den Verpackungstests kann es sich um Kartonfestigkeit und Etikettenhaftung handeln, Und Tertiär- Die Verpackung verwendet ISTA/ASTM-Testprotokolle. Alles muss im Validierungsplan dokumentiert werden.
Wichtige Schritte im Verpackungsvalidierungsprozess
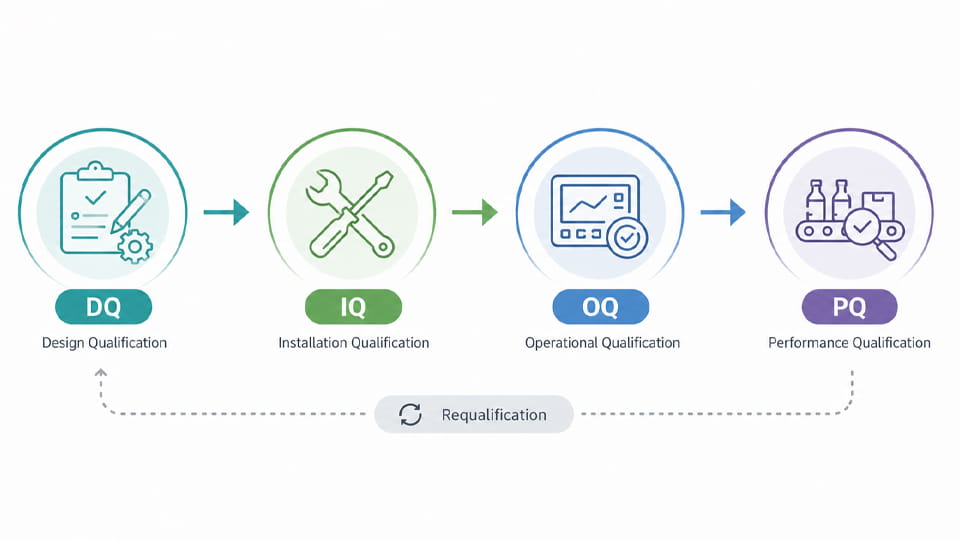
Die Verpackungsvalidierung folgt in der Regel dem Lebenszyklus der Geräte-/Prozessqualifikation, oft beschrieben als DQ/IQ/OQ/PQ. Diese Phasen stellen sicher, dass eine neue oder geänderte Verpackungslinie in jeder Phase den Anforderungen entspricht.
Designqualifikation (DQ): Frühe Phase, in der der Entwurf anhand des URS bewertet wird (Spezifikation der Benutzeranforderungen) und GMP-Richtlinien. Alle Designaspekte (Maschinenfunktionen, Materialien, Reinigbarkeit, Kompatibilität mit Produkt-/Verpackungsmaterialien) werden überprüft. Zu den Dokumenten gehören die URS und Designspezifikationen (Funktionale Designspezifikation FDS, Hardware-/Software-Designspezifikationen). Oftmals eine Gefährdungsbeurteilung (z.B. DFMEA) wird durchgeführt, um Verpackungsfehlermodi zu antizipieren.
Installationsqualifikation (IQ): Überprüft, ob die Maschine/Linie gemäß den Herstellerspezifikationen korrekt installiert ist. IQ umfasst eine Checkliste zur Überprüfung des korrekten Standorts, Dienstprogramme (Leistung, Luft, Wasser), Umgebungsbedingungen, und ordnungsgemäße Montage. Alle Komponenten sind vorhanden und unbeschädigt. Die Kalibrierung von Messgeräten wird überprüft. Leistungen: IQ-Protokolle und -Bericht, Kalibrierzertifikate.
Betriebsqualifikation (OQ): Überprüft, ob die Maschine unter allen angegebenen Bedingungen wie vorgesehen funktioniert. Kritische Parameter (Geschwindigkeitsbereiche, Temperaturen, Drücke, Vakuumniveaus, Drehmoment, usw.) werden bei Min/Max- oder Worst-Case-Einstellungen herausgefordert. Tests in der OQ könnten das Abdichten bei extremen Geschwindigkeiten umfassen, Sensor-/Alarmfunktionen, Fördergeschwindigkeit ändert sich, Genauigkeit des Etikettierers, Genauigkeit der Füllpumpe. Das OQ-Protokoll dokumentiert, dass jeder Funktionstest die Akzeptanzkriterien erfüllt.
Leistungsqualifizierung (PQ): Zeigt dies unter normalen Produktionsbedingungen, Die Linie produziert kontinuierlich Produkte, die den Qualitätsspezifikationen entsprechen. Die PQ erfolgt anhand tatsächlicher Produktionsmengen/Chargen. Dazu gehört der Betrieb der Linie für eine definierte Zeit oder Menge und die Probenahme der Ausgabe. Typische Aktivitäten: Betrieb mehrerer Blisterkartons, Flaschen bzw Beutel und Durchführung von QC-Tests an Proben (Z.B., Integrität des Behälterverschlusses (CCIT), Gewichtskontrollen, visuelle Mängelprüfung, Siegelfestigkeit). PQ zeigt, dass die Linie unter Beibehaltung der Produktqualität kontinuierlich arbeiten kann.
Die folgende Tabelle fasst die DQ-IQ-OQ-PQ-Stufen zusammen:
Bühne
Zweck
Schlüsseldokumente
Beispielaktivitäten/Tests
Designqualifikation (DQ)
Stellen Sie sicher, dass das Design des Verpackungssystems URS/GMP entspricht
Wie das Diagramm zeigt, Die Verpackungsvalidierung ist ein Lebenszyklusprozess vom Design bis zur Leistung. Beachten Sie, dass Umqualifizierung kann nach größeren Änderungen oder in regelmäßigen Abständen erforderlich sein.
Schlüsseltests in der Verpackungsvalidierung
Bei der Verpackungsvalidierung werden üblicherweise mehrere spezielle Tests durchgeführt:
Prüfung der Integrität des Behälterverschlusses (CCIT)
CCIT ist für sterile und sogar nicht sterile Produkte unerlässlich. Dabei wird beurteilt, ob das Behälterverschlusssystem (z.B. Fläschchen+Stopfen+Kappe, Ampulle, Blisterverschluss) bildet eine perfekte Barriere. Zu den Methoden gehört der Vakuumzerfall, Druckabfall, Eindringen von Farbstoff, oder mikrobielle Herausforderung. Wie eine Branchenquelle erklärt, „Prüfung der Integrität des Behälterverschlusses (CCIT) ist ein Test, der die Eignung von Behälterverschlusssystemen bewertet, um eine sterile Barriere gegen potenzielle Kontaminanten aufrechtzuerhalten.“. Regulierungsdokumente (USP <1207>, FDA, EMA-Anhang 1) Legen Sie Wert darauf, die Integrität des Verschlusses zu prüfen, um sicherzustellen, dass keine Undichtigkeiten oder Brüche auftreten. Zum Beispiel, USP <1207> bietet Richtlinien zur Auswahl von Dichtheitsprüfmethoden zur Zertifizierung versiegelter Verpackungen.
Prüfung der Materialverträglichkeit (Extrahierbare/auslaugbare Stoffe)
Verpackungsmaterialien (Kunststoffe, Gummis, Tinten, Klebstoffe) Es dürfen keine negativen Wechselwirkungen mit dem Arzneimittel auftreten. Extrahierbare und auslaugbare Stoffe Studien simulieren Langzeitkontakte: Extrahierbare Stoffe sind Chemikalien, die unter rauen Bedingungen aus der Verpackung austreten, und auslaugbare Stoffe sind solche, die unter normalen Bedingungen tatsächlich in das Produkt wandern. Diese Tests stellen sicher, dass die Verpackung keine giftigen oder die Stabilität beeinträchtigenden Verunreinigungen enthält. Pharmakopöe- und FDA-Leitlinien (z.B. USP <661>, Q3E) Überblick über die E/L-Prüfung für Behälterverschlusssysteme. In der Praxis, Man prüft, ob das Polymer vorhanden ist, Beschichtungen, und Etikettenmaterialien entsprechen den Arzneibuchstandards (z.B. USP-Glastyp, Spezifikationen für ISO-Gummiverschlüsse). Die WHO weist darauf hin, dass Arzneibuchstandards für Verschlüsse gelten (wie Gummistopfen) Sind „Mindestanforderungen“ und legt Wert auf Stabilitätsstudien zum Nachweis der Eignung.
Stabilitätsprüfung in der Verpackung
As per ICH Q1A, Stabilitätsstudien muss am Arzneimittel in seiner endgültigen Verpackungskonfiguration erfolgen. Während der Validierung, Der Einfluss der Verpackung auf die Stabilität wird bewertet: Zum Beispiel, Die Endverpackung wird einer beschleunigten Alterung unterzogen (hohe Temperatur/Luftfeuchtigkeit) und Echtzeitbedingungen, um sicherzustellen, dass keine Verschlechterung auftritt (z.B. Eindringen von Feuchtigkeit in Blisterverpackungen, Sauerstoffpermeation in Flaschen). Stabilitätsanzeigende Tests (chemisch und mikrobiell) durchgeführt werden. Eine Verpackung, die das Produkt unter Belastung nicht schützt, macht Angaben zur Haltbarkeit ungültig. Daher, Die Validierung umfasst die Bestätigung, dass die ausgewählte Verpackung die Haltbarkeitsanforderungen erfüllt (Dies überschneidet sich mit regulatorischen Stabilitätsprotokollen).
Transport- und Vertriebstests
Tertiärverpackung Tests simulieren tatsächliche Versandbedingungen. Zu den Standardtests gehören Fallhöhentests, Vibration (LKW-/Schienensimulation), Kompression (Stapelgewicht), und Temperaturwechsel (insbesondere für die Kühlkette). Zum Beispiel, ASTM D4169 (für verpackte Produkte) oder ISTA-Protokolle können verwendet werden. Ein robuster Verpackungsvalidierungsplan überprüft die Kartons, Kisten und Kisten schützen Primärverpackungen während der Handhabung: Stellen Sie sicher, dass kein Bruch vorliegt, Etikett löst sich ab, oder unter Verteilungsbedingungen treten Feuchtigkeitsschäden auf. Für die Kühlkette, Temperaturkartierung und validierte isolierte Transportbehälter werden getestet, um die erforderlichen Temperaturbereiche während des gesamten Transports einzuhalten.
Regulatorische Standards und Richtlinien
Die Validierung pharmazeutischer Verpackungen muss zahlreiche regulatorische Erwartungen erfüllen:
cGMP-Vorschriften: In den USA, FDAs 21 CFR Teil 211 (Unterabschnitt J) regelt Behälter und Verschlüsse. Abschnitt 211.94(Anzeige) legt fest, dass Behältnisse/Verschlüsse die Qualität des Arzneimittels nicht verändern dürfen und vor vorhersehbarer Kontamination schützen müssen.
EU-GMP: Europäische Richtlinien (EudraLex Vol. 4) verlangen das „Behälter sollten mit entsprechend validierten Methoden verschlossen werden“ (Annektieren 1) und dass Verpackungsprozesse GMP folgen (Annektieren 15 zur Validierung). EU-GMP-Anhang 11 schreibt eine computergestützte Systemvalidierung vor (einschließlich Verpackungslinien mit elektronischer Steuerung).
WHO GMP: Technische Berichtsreihe der WHO 902 (Annektieren 9) weist ausdrücklich darauf hin, dass Verpackungsgeräte wie jeder andere Verarbeitungsschritt einer Validierung bedürfen. Die WHO bietet auch Leitlinien zur Verpackungsintegrität (similar to EU/ICH).
ICH Guidelines: Obwohl ICH (F7, F8, F9, F10) kein spezifisches Verpackungsvalidierungsdokument haben, Sie schreiben ein Risikomanagement vor (F9) und Qualität durch Design (Q8/Q10) Grundsätze, die gelten. Zum Beispiel, Risikobewertung (ICH Q9) sollte Verpackungsattribute abdecken (Verschlusstyp, Etikettenkontrolle) die sich auf Produkt-CQAs auswirken.
Standards: Für sterile Produkte, ISO 11607 (für endsterilisierte Verpackungen medizinischer Geräte) wird oft analog verwendet; Teil 2 von ISO 11607 definiert Validierungsanforderungen für Form-/Siegelprozesse, Dies gilt für Blisterpackungen oder Ampullen für parenterale Arzneimittel. Pharmakopöen (USP, Ph. Eur.) enthalten auch Kapitel zu Verpackungstests (z.B. USP <381> für Gummiverschlüsse, <1207> zur Dichtheitsprüfung).
Zusammenfassend, Jede Verpackungslinie muss den nationalen Arzneibüchern und regionalen GMP-Standards entsprechen. In Compliance-Checklisten wird in der Regel die FDA zitiert, EU-GMP, WHO und relevante ISO-Standards, und fordern oft vollständig dokumentierte SOPs und Validierungs-Masterpläne für die Verpackung.
Häufige Herausforderungen bei der Verpackungsvalidierung
Die Verpackungsvalidierung stellt im Vergleich zu Herstellungsprozessen besondere Herausforderungen dar:
Datentyp und Stichprobe: Der Erfolg einer Verpackungslinie wird oft bestimmt durch diskrete Ergebnisse (z.B. Bestehen/Nichtbestehen für ein Siegel, Träne) statt kontinuierlicher Messungen. Wie ein Experte feststellt, „Die Art der erhaltenen Daten … stellt eine erhebliche Herausforderung dar. Über Erfolg oder Misserfolg entscheiden oft Mängel (Tränen, Löcher, Flecken, oder undichte Dichtungen)… Dieser Mangel an Messbarkeit (Variable) Daten erfordern oft sehr große Stichprobengrößen, damit ein Verpackungsprozess statistisch validiert werden kann.“. Mit anderen Worten, Möglicherweise benötigen Sie viele Proben, um die Zuverlässigkeit nachzuweisen.
Technologievielfalt: Es gibt viele Verpackungstechnologien (Blasenbildner, Kartonierer, Etikettierer, Kofferpacker, usw.) von verschiedenen Lieferanten. Jedes hat unterschiedliche Risikoprofile, Dies macht einen einheitlichen Validierungsplan unmöglich.
Änderungskontrolle: Verpackungslinien verarbeiten oft viele Produkttypen (Tabletten, Flüssigkeiten, Pulver) und Packungsgrößen auf der gleichen Ausrüstung (Verwendung von Änderungsteilen oder Formaten). Jede Änderung kann eine erneute Validierung oder Klammerung im Validierungsplan erfordern. Das managen (Die Validierungsmatrix) kann komplex sein.
Umweltkontrollen: Etwas Verpackung (z.B. Blisterverpackung feuchtigkeitsempfindlicher Arzneimittel) erfordert eine kontrollierte Luftfeuchtigkeit oder eine inerte Atmosphäre. Die Validierung dieser Kontrollen erhöht die Komplexität.
Integration mit Upstream-Prozess: Die Verpackungsvalidierung hängt von der Upstream-Konsistenz ab. Wenn die Granulatgröße des Medikaments unterschiedlich ist, Die Umstellung der Verpackungsmaschine kann davon betroffen sein (z.B. Marmelade füllen). Diese gegenseitige Abhängigkeit kann die Ursachenanalyse von Verpackungsfehlern erschweren.
Serialisierung/Tracking: Moderne Vorschriften (z.B. DSCSA, EU FMD) erfordern Serialisierung und Rückverfolgbarkeit. Sicherstellen, dass automatisierte Linien jede Einheit ordnungsgemäß verfolgen (und Validierung dieser Datenerfassung) fügt eine zusätzliche Qualifikationsebene hinzu (Softwarevalidierung, Barcode-Scanner-Tests, Datenbankprüfungen).
Regulatorische Unklarheit: Paradoxerweise, Es gibt nur begrenzte explizite FDA/EMA-Richtlinien ausschließlich zur „Verpackungsvalidierung“.,„Daher müssen Unternehmen häufig allgemeine GMP- und Prozessvalidierungsrichtlinien für Verpackungen interpretieren. Dies kann zu Unsicherheit über den genauen Umfang oder die erforderlichen Tests führen.
Trotz dieser Herausforderungen, das Grundprinzip bleibt bestehen: Behandeln Sie Verpackungen wie jeden anderen kritischen Prozess, mit gründlicher Qualifizierung und robusten QC-Kontrollen. So kommt das ISPE-Papier zu dem Schluss, Verpackungsvalidierung "abweichen(S) wenig von der Validierung von Prozessen zur Arzneimittelherstellung.“.
Best Practices für die Validierung pharmazeutischer Verpackungen
Um Herausforderungen zu meistern und eine erfolgreiche Validierung sicherzustellen, Befolgen Sie diese Best Practices:
Frühzeitige Risikobewertung: Nutzen Sie Qualitätsrisikomanagement (ICH Q9) aus der Designphase. Identifizieren Sie kritische Verpackungsattribute (z.B. Sterilität, Manipulationsnachweis, Lichtschutz) und priorisieren Sie Tests entsprechend. Ein Fehlermodus & Wirkungsanalyse (FMEA) wird während der DQ empfohlen, um den Validierungsumfang zu planen.
Benutzeranforderungen spez (URS): Dokumentieren Sie alle Anforderungen im Voraus klar und deutlich: Produktionskapazität, Packformate, Integrationspunkte (z.B. Anbindung einer Blistermaschine an einen Kartonierer), Umgebungsbedingungen, Reinigungsanforderungen. Ein gut definiertes URS sorgt dafür, dass die Validierung auf Kurs bleibt.
Standardarbeitsanweisungen (Sops): Pflegen Sie detaillierte SOPs für den Gerätebetrieb, Reinigung, Umstellung, und Wartung. Die Validierung sollte sich auf diese SOPs beziehen, um eine konsistente Verwendung sicherzustellen.
Automatisierte Inspektion und Kontrollen: Integrieren Sie Inline-Qualitätsprüfungen (z.B. Bildverarbeitungssysteme für die Druck-/Etiketteninspektion, Kontrollwaagen, Barcode-Scanner, Lecksucher). Dabei handelt es sich nicht nur um QS-Tools, sondern auch um einen Teil der Validierung durch die Erkennung von Fehlern in Echtzeit.
Umfangreiche Dokumentation: Bereiten Sie einen Validierungs-Masterplan vor, in dem die Strategie dargelegt wird, und stellen Sie alle Qualifizierungsprotokolle sicher (IR/WH/PQ) klare Akzeptanzkriterien haben. Führen Sie detaillierte Protokolle aller Läufe, Abweichungen, und erneute Tests. Nutzen Sie Checklisten, um sicherzustellen, dass nichts übersehen wird.
Ausbildung und Personal: Bediener und QC-Personal müssen sowohl in Bezug auf die Ausrüstung als auch auf die Validierungsverfahren geschult werden. Nur qualifiziertes Personal sollte Validierungstests durchführen und Ergebnisse aufzeichnen.
Software und elektronische Aufzeichnungen: Für computergestützte Steuerungen, Folgen Sie der FDA 21 CFR Teil 11/GMP-Anhang 11: Software validieren, Audit-Trails, elektronische Signaturen (sorgen für Sicherheit, Backups). Selbst nicht sterile Verpackungslinien verfügen häufig über SPS/HMIs, die validiert werden müssen.
Design zur Validierung: Wann immer möglich, Wählen Sie Maschinen mit Funktionen, die die Validierung vereinfachen: z.B. Servoantriebe mit digitaler Steuerung (für präzise, reproduzierbare Parameter), hygienische Edelstahlrahmen (für eine einfache Reinigung), leicht austauschbare Teile (um den Reinigungsaufwand zwischen den Formaten zu reduzieren), und modulares Design (um die Anzahl der Konfigurationen zu begrenzen, die getestet werden müssen).
Laufende Überprüfung: Die Verpackungsvalidierung ist nicht „einmal erledigt“. Implementieren Sie regelmäßige Überprüfungs- oder Revalidierungsauslöser (z.B. nach größeren Wartungsarbeiten, Ausrüstungs-Upgrades, oder erhebliche Prozessdrift). Nutzen Sie die statistische Prozesskontrolle (SPC) zu wichtigen Kennzahlen (Variation des Füllgewichts, Fehlerrate) um Trends frühzeitig zu erkennen.
Datenintegrität: Wenden Sie die ALCOA+-Prinzipien an: alle Validierungsdaten (Prüfprotokolle, QC-Berichte, Kalibrierungen) muss zurechenbar sein, lesbar, gleichzeitig, Original, und genau. Verwenden Sie gebundene Notizbücher oder validierte elektronische Systeme.
Durch die Einbindung von Validierungsdenken in jeder Phase – vom Design bis zum täglichen Betrieb – können Hersteller sicherstellen, dass Verpackungslinien Produkte liefern, die den Qualitäts- und Compliance-Erwartungen entsprechen.
Rolle von Verpackungsmaschinen bei der Validierung
Die Wahl und Gestaltung der Verpackungsausrüstung hat großen Einfluss auf den Validierungserfolg. Hochwertige Verpackungsmaschinen sind so konstruiert, dass sie den GMP-Anforderungen entsprechen und eine einfache Qualifizierung ermöglichen. Zum Beispiel, Jinlu Packing Automatisierte Maschinen sind mit Funktionen ausgestattet, die die Validierung optimieren:
Präzision und Konsistenz: Jinlu Blasenmaschinen, Kartonierer und Füller verwenden Servomotoren und präzise Mechanik, um einen gleichmäßigen Heißsiegeldruck sicherzustellen, Volumen füllen, und Indizierung. Diese Wiederholbarkeit ist entscheidend für das Bestehen von OQ- und PQ-Tests (z.B. Die servoangetriebene Vorschubeinrichtung sorgt dafür „genaue Positionierung“ auf ihrer Blasenlinie).
Hygienisches Design: Materialien und Oberflächen sind Edelstahl oder FDA-konforme Kunststoffe, Entwickelt für eine einfache Reinigung. Glatt, Gefälleflächen verhindern Staubeinschlüsse. Dies unterstützt die GMP-Konformität und vereinfacht die Reinigungsvalidierung.
Anpassungsfähigkeit an Formate: Jinlu bietet kundenspezifische Zuführungen und Führungsschienen für verschiedene Pakettypen an (sehen Benutzerdefinierte Feeder auf Produktseiten). Mit den richtigen Werkzeugen werden Abweichungen reduziert und Formatwechsel beschleunigt, Dadurch verringert sich der Requalifizierungsbedarf beim Formatwechsel.
Integrierte Kontrollen: Moderne Jinlu-Linien verfügen über SPS/HMI-Steuerungen, die elektronische Chargenprotokolle erstellen können. Zum Beispiel, Die Flüssigkeitsfüllmaschine verfügt über eine KI Siemens SPS/HMI für einfach, nachvollziehbarer Betrieb. Solche digitalen Steuerungen unterstützen die Datenprotokollierung (im Einklang mit 21 CFR Teil 11/Anhang 11) – entscheidend für die Validierung, dass Sollwerte und Alarme wie vorgesehen funktionieren.
Validierungsdokumentation: Jinlu stellt eine vollständige Dokumentation zur Verfügung (Datenblätter, Handbücher, usw.). Zum Beispiel, ihre Produktseitenliste „Vollständiger Satz technischer Dokumente“ und sie legen Wert auf die Bereitstellung detaillierter Design- und Testdokumentationen. Ein Lieferant, der bereit ist, einen Werksabnahmetest durchzuführen (FETT) Protokolle oder IOQ-Kits erleichtern die Validierung des Benutzers erheblich.
Qualitätszertifizierungen: Jinlu-Tragemaschinen CE, cGMP und andere Zertifizierungen, Dies weist darauf hin, dass sie bestimmte Qualitäts- und Sicherheitsstandards erfüllen (siehe das cGMP-Logo auf den Produktspezifikationsbildern). Der Einsatz zertifizierter Geräte kann die behördliche Überprüfung rationalisieren.
In der Praxis, Eine validierte Verpackungslinie könnte Folgendes umfassen:: ein Jinlu Blisterverpackungsmaschine verknüpft mit a Kartoniermaschine Und Zähl-/Fülllinien. Jedes Gerät muss qualifiziert sein (IR/WH/PQ). Die Systeme von Jinlu sind so konzipiert, dass sie nahtlos zusammenarbeiten (z.B. „hochautomatisierte Blisterkartonierlinie“ bis zu 320 Kartons/Min), Reduzierung von Integrationsproblemen während der Validierung. Wir bieten auch Fallstudien an (z.B. eine vollständige Zähl- und Abfülllinie, oder ein automatische Kartonierung & Beschriftungszeile) die durchgängig validierte Lösungen demonstrieren.
DPH-270Max Rollenblisterverpackungsmaschine
Durch eine Partnerschaft mit Jinlu oder ähnlichen Herstellern, Unternehmen profitieren von der Ausrüstung „cGMP-konform“, unterstützt einen vollständigen Validierungslebenszyklus, und kommt mit After-Sales-Support (Inbetriebnahme, Ausbildung) um sicherzustellen, dass die Leitung validiert bleibt.
Abschluss
Verpackungsvalidierung ist für die pharmazeutische Qualität und Compliance von entscheidender Bedeutung. Indem Verpackungsvorgänge genauso streng behandelt werden wie zentrale Herstellungsschritte, Unternehmen stellen sicher, dass ihre Produkte sicher bei Patienten ankommen. Der Prozess umfasst die Definition (URS, Risikobewertung), Qualifikation (DQ/IQ/OQ/PQ), Testen (CCIT, Stabilität, Transport), und kontinuierliche Überwachung. Es unterliegt den GMP-Vorschriften (FDA, EMA, WER) und Industriestandards.
Moderne automatisierte Verpackungsanlagen – wie die Maschinen von Jinlu Packing – spielen bei der Validierung eine Schlüsselrolle. Auf Präzision ausgelegt, Hygiene und elektronische Rückverfolgbarkeit, Sie helfen Herstellern, Validierungskriterien effizient zu erfüllen. Bei der Auswahl von Verpackungslinien, Ziehen Sie Lieferanten in Betracht, die eine vollständige Validierungsdokumentation und Unterstützung bereitstellen.
Letztlich, Eine gründliche Verpackungsvalidierung ist erforderlich „der letzte Garant für Produktqualität und Patientensicherheit“, Ausrichtung sowohl an regulatorischen Anforderungen als auch an Geschäftszielen.
FAQs zur Verpackungsvalidierung in der Pharmaindustrie
Was ist der Unterschied zwischen Verpackungsvalidierung und Prozessvalidierung??
Die Prozessvalidierung bezieht sich typischerweise auf die Schritte zur Herstellung der Arzneimittelsubstanz oder des Massenarzneimittelprodukts (z.B. mischen, Granulation, Kompression). Bei der Verpackungsvalidierung werden Validierungsprinzipien speziell auf die Verpackungsvorgänge angewendet (Versiegelung, Beschriftung, Kartonfüllung, usw.). Jedoch, Beide folgen demselben DQ/IQ/OQ/PQ-Lebenszyklus und denselben GMP-Anforderungen. Im Wesentlichen, Die Verpackungsvalidierung stellt den Verpackungsprozess sicher (nicht nur der Herstellungsprozess) erfüllt stets die Qualitätsanforderungen.
Welche Tests sind für die Validierung von Arzneimittelverpackungen erforderlich??
Zu den wichtigsten Tests gehört die Integrität des Behälterverschlusses (CCIT) um Siegel zu überprüfen, Materialkompatibilität (Studien zu extrahierbaren/auslaugbaren Stoffen), Stabilitätstests (Medikament in Endverpackung unter Stress), und Verteilungstests (Schock, Vibration, Temperatur für den Transport). Zusätzliche Prüfungen betreffen die Genauigkeit der Etiketten, visuelle Inspektionssysteme, Gewicht/Kontrollwägung, und etwaige vertragsspezifische Tests (z.B. sterile Filmstärke). Alle Tests sollten vordefinierte Akzeptanzkriterien haben und dokumentiert werden.
Wie lange dauert die Verpackungsvalidierung normalerweise??
Der Zeitplan hängt von der Komplexität ab (Anzahl der Formate, Ausrüstung, Websites). Ein einzeiliger IQ/OQ/PQ kann zwischen einigen Wochen und einigen Monaten liegen. Faktoren: die Anzahl der OQ-Parametertests, erforderliche Stichprobengrößen in PQ, und Zeit für die Testdurchführung (z.B. Die Stabilität unter beschleunigten Bedingungen dauert Wochen). Planen paralleler Aktivitäten (wie das Vorbereiten von Protokollen, während die Ausrüstung eintrifft) kann Zeit sparen.
Welche Dokumente regeln die Verpackungsvalidierung??
Die primären Referenzen sind GMP-Vorschriften: 21 CFR Teil 211 (US-amerikanische FDA), EudraLex Vol. 4 (EU-GMP, Annektieren 1 Und 15), und WHO-GMP-Anhang 9. ICH Q7/Q10 bieten allgemeine Validierungsprinzipien, ICH Q9 deckt das Risikomanagement für Verpackungsentscheidungen ab, und ISO 11607-2 Gilt für die Prozessvalidierung von Sterilverpackungen. Arzneibuchstandards (USP, Ph. Eur.) und lokale Richtlinien (z.B. Chinesisches NMPA, Indien Zeitplan M) beeinflussen auch Verpackungsanforderungen. Halten Sie sich immer an die Vorschriften des Zielmarktes.
Wie unterstützt Jinlu Packing die Verpackungsvalidierung??
Die Verpackungsmaschinen von Jinlu sind GMP-konform gebaut. Sie stellen Konstruktionsunterlagen zur Verfügung (URS, FDS), Kalibrierzertifikate, und OQ/PQ-Unterstützung. Ihre Ausrüstung (Blasenmaschinen, Kartonierer, Fülllinien) Verfügt über eine präzise Servosteuerung und ein hygienisches Design, um die Validierungsspezifikationen zu erfüllen. Wie auf Jinlus Website gezeigt, Maschinen tragen cGMP/CE-Logos und werden mit technischen Dokumentationssätzen geliefert. Jinlu bietet auch Installations- und Validierungsunterstützung, individuelle Lösungen, und Schulungen zur Unterstützung bei der Implementierung validierter Verpackungslinien. Weitere Informationen, Weitere Informationen finden Sie auf den Seiten zu Blisterverpackungsmaschinen oder Kartoniermaschinen von Jinlu.
Kleines Fu, Gründer von Jinlupacking, bringt vorbei 20 langjährige Erfahrung im pharmazeutischen Maschinenbau. Unter seiner Führung, Jinlu hat sich zu einem vertrauenswürdigen Lieferanten für integriertes Design entwickelt, Produktion, und Verkäufe. Petty teilt mit Leidenschaft sein umfassendes Branchenwissen, um Kunden bei der Bewältigung der Komplexität von Pharmaverpackungen zu unterstützen, Sicherstellen, dass sie nicht nur Ausrüstung erhalten, sondern eine echte, auf ihre Produktionsziele zugeschnittene Servicepartnerschaft aus einer Hand.