
で 医薬品製造, 確保する 機器サプライヤー GMP基準を満たすことは、原材料ベンダーを精査するのと同じくらい重要です. 徹底した サプライヤー監査 製薬会社が機器メーカーの品質システムを検証するのに役立ちます, ドキュメント, とプロセス, 欠陥のある機械や規格に準拠していない機械が生産に入るリスクを軽減します。. 規制に関するガイダンス (FDA, エマ, 私, 誰が) を強調します リスクベースのサプライヤー品質プログラム: 重要なベンダーのサプライヤー認定監査を実施する, 品質管理システムとコンプライアンス履歴をレビューする, 是正措置と承認をフォローアップします.
この記事では、機器サプライヤーを監査する理由とタイミングについて説明します。, 段階的な監査プロセスの概要を示します (フローチャート付き), 機器に焦点を当てた監査チェックリストを提供します, オンサイトと比較. リモート監査, FAT/SAT と IR/WH/PQ サプライヤー資格に適合する. 表は監査の種類とリスクレベルをまとめたものです, そして実践的なアドバイス (実際の例と規制上の引用を含む) 調達に役立ちます, QA チームとエンジニアリング チームが監査を成功裏に実行. 要するに, 構造化された監査プログラム - によってサポートされています。 包括的なドキュメント GMP に精通したサプライヤーからの提供 - 新しい機械が IQ/OQ 認定および GMP 使用のために「監査準備完了」状態で到着することを保証します.

規制された製薬会社で, 何も偶然に任せることはできません. たとえ機械が良く作られていても、, サプライヤーにおける品質上の欠陥 (貧弱なQMS, 文書化されていない変更, 校正されていない機器, 等) 製品を危険にさらす可能性があります. なぜなら製薬メーカーは、 最終的に責任がある 最終製品の品質とコンプライアンスのために, 規制当局は重要なサプライヤーを監査することを期待している. 例えば, FDA の ICH Q10 ガイダンス 契約者はサプライヤーの「適合性と能力を評価する責任がある」と指摘している, すべての品質関連活動は書面による契約で定義されなければなりません. 実際に, これは、機器ベンダーが GMP のような管理に準拠していることを確認するために監査することを意味します。. ある業界専門家は次のように説明しています。, サプライヤー監査は、原材料を改善するための定期的な検査です。, 装置, 企業が受け取る供給品…そして厳格な規制要件へのコンプライアンスの達成を支援します.
機器サプライヤーを監査する主な理由は次のとおりです。:
「サプライヤー監査は、サプライヤーのプロセスや製品をベンチマーク基準に照らして独立して評価するもので、製品を改善する機会を明らかにすることを目的としています。, プロセス, またはシステム,」 質の高い業界情報源が説明する. 製薬業界で, これは、機器ベンダーの品質マニュアルを確認することを意味します。, 変更制御, キャパ, および文書はすべて GMP の期待を満たしています. 堅実な監査プログラム (初期および定期的) 機器がGMP条件下で安全に機能することを検証するのに役立ちます.

監査は主要な段階で計画する必要がある. 典型的なタイミングとしては次のものが挙げられます。:
以下の表は、一般的な監査の種類とその目的をまとめたものです。:
| 監査の種類 | 目的 |
| イニシャル (資格) | を評価する 新しい 承認前のサプライヤー; QMSを確認する, ドキュメント, と機能. |
| 定期的 (予定されている) | 既存のサプライヤーを再評価する (例えば. 毎年) 継続的なコンプライアンスを確保するために. |
| 理由のため (トリガー) | 特定の問題を調査する (例えば. 苦情, 逸脱, 483 観察) サプライヤーで. |
| フォローアップ | 是正措置/予防措置が講じられていることを確認する (キャパ) 以前の監査からの事項が実施されている. |
| リモート/デスク監査 | サプライヤーの品質システムとドキュメントを仮想的にレビューします; 低コスト/高速, ただし範囲が限られている. |
すべての場合において, を使用します リスクベースのアプローチ 範囲を決める. 高リスクのサプライヤー (重要な機器) より深い保証, 現場監査, 一方、低リスクのものは机上レビューまたはサンプリングのみが必要な場合があります. EMAのQ&A は、サプライヤーが GMP 証明書または検査報告書を持っている場合でも、, メーカーの法的責任は、独自の監査プログラムを実施するまで満たされない. 言い換えると, 外部評価を使用して優先リストを通知する, ただし、独自の監査を通じて信頼し、検証してください。.
シンプルなリスク マトリックスは、サプライヤーを分類し、監査頻度をガイドするのに役立ちます. 例えば:
| リスクレベル | 例 | 監査アクション |
| 高い | 無菌液体充填剤, 滅菌器, カプセル充填剤; 初めてのサプライヤーまたは単一ソースのサプライヤー | 徹底した初期現地監査; 年次または隔年監査; フルスコープ (QMS, 施設, 書類, 脂肪). |
| 中くらい | 非滅菌包装機 (例えば. 水ぶくれ, 箱詰め業者); セカンドベンダーまたは既知のサプライヤー | 詳細な初期監査; 2 ~ 3 年ごとの現場監査; 中程度の範囲 (重要なプロセスに焦点を当てる, プラスドキュメント). |
| 低い | 付帯機器 (例えば. コンベア, 基本的なポンプ); 商品ベンダー | 基本資格 (ドキュメントのレビュー, アンケート); 問題が発生した場合のみオンサイト監査; 定期的な再評価 (3–5年または必要に応じて). |
このリスク評価で考慮すべきことは、 インパクト (製品の安全性/品質にとって機器はどの程度重要ですか?) そして 可能性 (サプライヤーは過去に問題を抱えていましたか? 新しいデザイン? 複雑な技術?). 例えば, 無菌医薬品工場の新しいカプセル充填ラインは大きな影響を及ぼします (直接連絡, 高精度), そのため、高リスクのカテゴリーに留まり、完全な監査が必要となります。. 対照的に、, 製品に直接接触しない単純なコンベヤはリスクが低い可能性があります – 最初はリモート監査またはアンケートで十分かもしれません, リスクレベルが変化した場合のみオンサイトチェックを行う.
以下は監査プロセスの大まかなフローチャートです。, その後に各ステップの詳細が続きます:
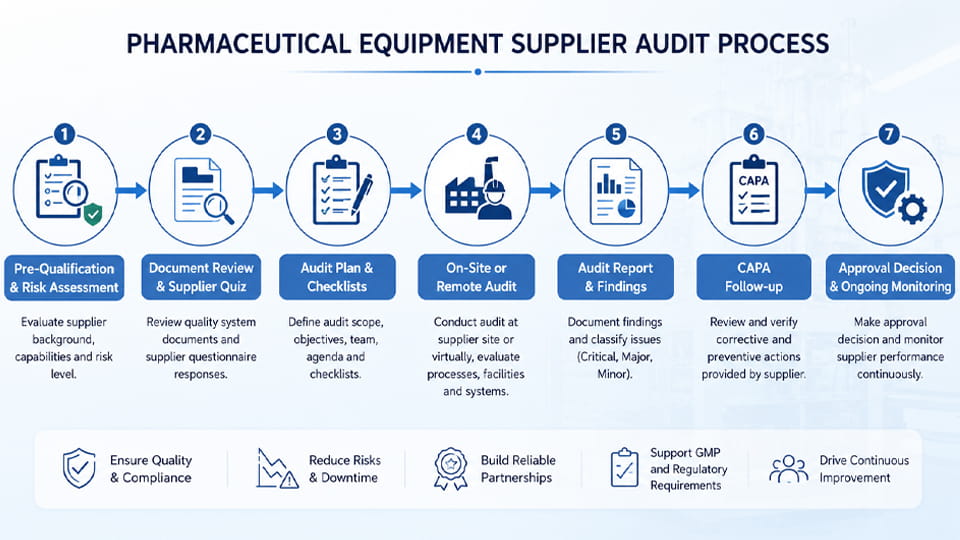
各ステップの概要を以下に示します:
を実施する場合 リモート監査, 書類審査に重点を置く, ライブビデオのウォークスルー, そしてインタビュー. EMA が指摘しているように, リモート監査は監査プログラムに情報を提供できます, しかし、リスクの高いサプライヤーに対する現地検査ほど徹底的ではありません。. 真に重要な機器については、常に完全なオンサイト監査を行うことを目指します.
以下は、機器サプライヤーの監査中に考慮すべき重要な領域と質問例をまとめたチェックリストです。. (これを特定のマシンとリスク レベルに合わせて調整してください。)
このチェックリストは出発点です. 特定の機器に合わせて調整する: 例えば, ある ブリスターパッカー 監査はヒーターの校正に焦点を当てます, 日付コーダ, そしてフィルムの取り扱い, その間 カプセルフィラー 監査では、線量の精度チェックとカプセルロックの検証が重視される. 目標は両方を評価することです 紙に書かれていること (手順, 記録) そして 床で何が起こっているのか (観察された実践).

機器サプライヤーを監査する場合, 最も頻繁に観察される問題には次のようなものがあります。:
これらのいずれかに遭遇した場合, サプライヤーと協力して是正計画を策定する. 最終承認の前に、監査結果を適切に解決することが重要です. アクションアイテムを常に記録し、フォローアップします。すべての重要な CAPA が検証されるまで、サプライヤーは認定されません。.
| 側面 | 現場監査 | リモート監査 |
| 範囲 | フルアクセス (設備, 人々, 書類). | 書類に限ります, 記録, およびバーチャルツアー. |
| 検査の深さ | 高い: 機械を物理的に検査できる, ハウスキーピング, 目に見えない実践. | 適度: 主に書類審査と面接. |
| 料金 & 時間 | より高い (移動時間と費用, スケジュール設定の課題). | より低い (旅行はありません; より迅速なスケジュール設定). |
| 効果 | 最も徹底した (リスクの高いサプライヤーに好まれる). | フォローアップまたはリスクの低いベンダーに適しています; 緊急のニーズに対する暫定的なソリューション. |
| 柔軟性 | 柔軟性が低い (スケジュール/空き状況を同期する必要がある). | より柔軟に (多くの場合、ビデオ経由で複数のサイトを監査できます). |
| 監査証拠 | プロセスの直接観察, 写真/ビデオ; 不一致を見つけやすくなる. | サプライヤーが示すものに依存する; 隠れた問題を見逃すリスク. |
一般的に, 現場監査 ゴールドスタンダードであり続ける, 特にリスクの高いサプライヤーまたは新たに資格を取得したサプライヤーの場合. 規制当局は、遠隔地からの「紙ベースの」監査は現場での監査の完全な代替にはならないと指摘. しかし, リモート監査は役立つ場合があります (例えば, 旅行が制限されている場合の一時的な措置として) サプライヤーの品質記録を効率的にレビューできます. ハイブリッドアプローチが一般的: 基本的な文書レビューにリモート監査を使用する, その後、最終的な詳細調査のために現地訪問をスケジュールします。, または、定期的なチェックのためにオンサイトとリモートを交互に行います。.
機器監査では次の点に特に注意を払う必要があります。 検証ドキュメント. 購入する前, サプライヤーが堅牢な FAT 手順を備えていることを確認する. 完全な FAT により、制御された条件下でマシンが URS に対して動作することが検証されます。. 監査中, 同様のマシンに関するサプライヤーの過去の FAT レポートを確認する: テスト基準と結果が明確に文書化されており、仕様を満たしていることを確認します。. また、インストール後に正式な SAT 計画が実行されることを確認します。. これらの受け入れテストは資格を得るために重要です.
同じく, サプライヤーが完全な IQ/OQ サポートを提供していることを確認する. Jinlu の経験は、信頼できるベンダーが機械を提供していることを示しています 「完全な IQ/OQ/PQ サポートとターンキー検証サービス付き」. 実際に, これは、IQチェックリストを提供する必要があることを意味します, OQ テスト スクリプト, レビュー用のサンプル PQ プロトコル. ベンダーは FAT を実行することが多く、制御された条件下でマシンを事前に調整して設置することで IQ を支援できます。. でも覚えておいてください: の 最終的な責任は購入者の QA チームにあります オンサイトで IQ/OQ/PQ を完了するため. 監査では、サプライヤーが検証をサポートできる訓練を受けたスタッフまたは請負業者を配置し、必要な報告書を提供することを確認する必要があります。, 図面, および校正記録.
直接質問する, 例えば: 「当社の URS に基づいた FAT プロトコルはありますか??」, 「現場でIQ/OQテストを目撃しませんか?」, その後、検証済みのドキュメントを提供します?」. 優れたサプライヤーはテンプレートを用意しており、これを日常的なものとして扱います。これは GMP 対応の一環です。.
監査中 (または監査前の議論), 買い手はよくサプライヤーに尋ねます:
これらに対する答えは、サプライヤーの成熟度を評価するのに役立ちます. 例えば, 経験豊富なサプライヤーはすぐに QA 証明書を提示する必要があります (ISO 9001/13485) 検証プロセスの詳細を説明するのに問題はありません. サプライヤーがこれらの質問をためらったり、そらしたりした場合, システムのギャップを示している可能性があります.
監査と回答の収集後, 最後のステップはサプライヤーの承認です. すべての監査結果が終了するか解決されたら, 品質システムでサプライヤーを正式に承認する. これには、承認ベンダー リストへの設定や条件の指定が含まれる場合があります。 (例えば, 「初回注文には SAT 後に QA による承認が必要です」). この承認を調達チームとプロジェクト チームに伝えます, サプライヤーが適格であることがわかるようにするため. サプライヤーが残りの期待を理解していることを確認する (今後のレポートで最新の CAPA 証拠やパフォーマンス指標を提供するなど).
承認後も, 監視を続ける. 主要なサプライヤー指標を追跡し続ける (時間通りの配達, 現場の問題, 監査スコア). 新しい FDA または EMA 要件を随時統合します (規制が変わったら, 再監査または新しい文書の要求が必要になる場合があります). 本質的には, 監査プログラムは承認にとどまりません。監査プログラムは、あらゆる重要な機器の継続的な品質保証の一部となります。.
構造化されたサプライヤー監査プログラムは、製品の安全性と規制の信頼性への投資です. 機器メーカーの品質システムを系統的にチェックすることにより、, ドキュメント, GMP 原則に基づいたプロセスにより、予期せぬ事態を最小限に抑え、準拠した機械のみがラインに投入されるようになります。. 覚えて: 共同責任 鍵です. サプライヤーは、完全な FAT/SAT/IQ/OQ パッケージを備えた GMP 対応マシンを持参する必要があります。, ただし、会社は生産前にそれらを検証する必要があります. リスクベースのアプローチにより、重要な箇所に重点的に取り組みます, そして、いかなる発見もCAPAと最終的な承認決定につながる必要があります.
ジンルーパッキングにて, 私たちはGMP監査の厳格さを理解しています: 当社の包装機は、これらのニーズを満たすように設計され、文書化されています。. 各マシンには完全な検証キットが付属しています (FAT/SAT/IQ/OQプロトコル) 資格取得を効率化するために. 製薬コンプライアンスに経験のあるサプライヤーを選択することにより, 監査を簡素化し、プロジェクトを順調に進めます.
自信を持って監査する準備ができています? ジンルパッキングにお問い合わせください 付属の製薬グレードの機器向け 監査対応. 私たちのチームが URS の定義をお手伝いします, FATを実行する, サプライヤーの監査と認定がスムーズに行われるように、ターンキー IQ/OQ/PQ ドキュメントを提供します.
サプライヤー監査は、製薬機器メーカーの品質システムの体系的な評価です。, 製造工程, 技術力, およびコンプライアンスの実践. 目標は、サプライヤーが GMP 要件を満たす機械を一貫して提供できることを検証することです。, 品質基準, そしてプロジェクトの仕様.
サプライヤー監査は製薬会社がプロジェクトのリスクを軽減するのに役立ちます, サプライヤーの能力を検証する, コンプライアンスの準備状況を評価する, 機器メーカーがGMP規制の生産に必要な文書化と検証サポートを提供できるようにする.
適切なサプライヤー監査がなければ, 購入者は検証の遅れなどの問題に直面する可能性があります, マシンのパフォーマンスが悪い, 不完全な文書, または規制遵守の課題.
重要な文書にはサプライヤーの品質マニュアルが含まれます, SOP (キャパ, 変更制御, 等), ISO/GMP認証, サンプル製品のバッチ記録, トレーニング記録, メンテナンス/校正ログ, および以前の監査報告書. 分析証明書 (CoA) 最近のバッチとサプライヤーの安定性データも重要です. 本質的には, 品質システムと製品リリース管理を実証するすべてをレビューする.
はい. ビデオ会議を使用してリモートのサプライヤー監査を実行できます, デジタルドキュメントのレビュー, バーチャル工場見学.
しかし, カプセル充填機などの重要な製薬機器用, 打錠機, ブリスター包装機, および無菌処理装置, 現場監査は、購入者が製造慣行を直接評価できるため、一般に推奨されます。, 品質管理, および施設の状況.
サプライヤー認定は、ベンダーを承認する全体的なプロセスです (アンケートを含む, 書類審査, サンプルテスト, そして監査). サプライヤー監査は、そのプロセスにおける特定のステップの 1 つです (または定期監視中). 資格認定には書類手続きと現場監査の両方が使用されます. 資格取得に成功した後は, 監査は定期的なチェックになる (再資格).
FDAとEMAはいずれも製薬会社がサプライチェーンを管理することを期待している. FDA の Q10 は、リスクベースのサプライヤー評価を強調し、特にサプライヤーの品質を検証するためのサプライヤー監査を求めています。. EU GMP (別館 16 およびパートIの章 7) 製品の製造またはテストに関与するすべてのサイトの監査を有資格者が利用できるようにすることを要求します. I Q7 (API用) および PIC/S ガイドラインも同様にサプライヤーの強力な監視を義務付けています. 要するに, 規制当局は、GMP コンプライアンスの一環としてサプライヤーを監査していると想定しています。.
頻度はサプライヤーのリスクと過去の実績に依存します. 高リスクのサプライヤー (API, 無菌コンポーネント) 年次監査を受けることが多い, 一方、リスクの低いサプライヤーはあらゆるリスクを抱えている可能性があります。 2-3 年. 大きな変更や品質イベントの後にも監査します. 品質管理システムでは、リスク カテゴリごとに特定の間隔を定義する必要があります。. サプライヤーにおける業績不振や安全性の低下に関するイベントは、スケジュールに関係なく「原因に基づく」監査が必要であることに留意してください。.
脂肪 (工場受け入れテスト) 機器の出荷前にサプライヤーの施設で実施される正式なテストです.
FAT はそれを検証します。:
• マシンはユーザーの要件を満たしています
• 重要な機能が正しく動作する
• 安全システムは期待どおりに動作します
• ドキュメントが完成している
FAT が成功すると、設置のリスクが軽減され、その後の認定作業が簡素化されます。.
いくつかの重要な質問には次のようなものがあります。:
• 製薬プロジェクトを何件完了しましたか?
• FATを提供してもらえますか?, 土, および IQ/OQ ドキュメント?
• 設計変更をどのように管理するか?
• あなたの会社はどのような認定を取得していますか?
• 顧客からの苦情と CAPA にどのように対処しますか?
• スペアパーツのサポートポリシーは何ですか?
• インストール後に利用できるトレーニング サービス?
• トレーサビリティと文書管理をどのように確保するか?
これらの質問は、買い手が技術的能力と長期的なパートナーシップの可能性の両方を評価するのに役立ちます.
購入者は評価する必要があります:
• エンジニアリングチームの経験
• 自動化の専門知識
• PLC および HMI プログラミング機能
• カスタマイズ経験
• GMP設計の知識
• 過去の製薬プロジェクト
• 検証サポートの経験
サプライヤーのエンジニアリング能力は、多くの場合、機械の信頼性に直接影響します。, スケーラビリティ, そして長期的なパフォーマンス.
参考文献:
1.ICH Q10 医薬品品質システム – 科学的ガイドライン — 欧州医薬品庁
2.Q10 医薬品品質システム — 私たち. 食品医薬品局
3.ユードラレックス – 音量 4 – 優れた製造業 (GMP) ガイドライン — 欧州委員会
4.VDI 6306 – 製薬業界におけるサプライヤー監査 – VDI ドイツ技術者協会 e.V.



