Na indústria farmacêutica, qualidade e segurança do paciente são fundamentais. Cada comprimido, frasco, ou blister deve ser produzido sob controles rigorosos para garantir que seja eficaz, puro, e seguro. É onde Conformidade com GxP entra. GxP é um termo genérico que significa “Bom x Prática,”onde “x” pode ser Fabricação, Laboratório, Clínico, Distribuição, etc.. Essencialmente, As diretrizes GxP são regulatórias sistemas de qualidade que as empresas farmacêuticas deve seguir documentar e controlar todos os processos. Junto, estas regras garantem que os medicamentos sejam consistentemente produzidos e controlados de acordo com padrões de alta qualidade ao longo do desenvolvimento, fabricação, teste, embalagem, e distribuição.
Reguladores como o FDA (NÓS. Food and Drug Administration), Ema (Agência Europeia de Medicamentos), QUEM, e outros impõem padrões GxP. Por exemplo, as Boas Práticas de Fabricação Atuais da FDA (Cgmp) regras (21 Peças CFR 210–211) definir requisitos mínimos para fabricação, embalagem, e guardar medicamentos para garantir que sejam seguros e tenham os ingredientes e a dosagem corretos. De forma similar, As diretrizes da OMS e da UE abrangem GMP/GLP/PIB/GVP globalmente. Na prática, empresas farmacêuticas desenvolvem Sistemas de Gestão da Qualidade e Procedimentos Operacionais Padrão (POPS) em torno dos princípios GxP. Cada lote de produto possui registros rastreáveis do início ao fim. Até os equipamentos devem ser qualificados (com qualificação de instalação/operacional/desempenho – IQ/OQ/PQ) e validado para atender aos requisitos GxP.

O que significa GxP em produtos farmacêuticos?
A sigla GxP significa Boa prática “x”, onde o “G” é “Bom” e o “P” é “Prática”. O “x” representa várias disciplinas. GxP não é um regulamento único, mas um grupo de diretrizes de qualidade. As principais disciplinas GxP incluem:
- Boas Práticas de Fabricação (BPF): Garante que os produtos sejam consistentemente produzidos e controlados de acordo com padrões de qualidade. GMP cobre fabricação, equipamento, instalações, pessoal, e documentação para minimizar a contaminação, confusões, e erros.
- Boas Práticas de Laboratório (BPL): Rege a realização de estudos laboratoriais não clínicos (como testes toxicológicos) para garantir a integridade e a reprodutibilidade dos dados. GLP garante que os testes de laboratório sejam bem documentados, auditável, e confiável.
- Boas Práticas Clínicas (GCP): O padrão internacional para projetar, conduzindo, monitoramento, e relatórios de ensaios clínicos. GCP protege os direitos dos pacientes, integridade dos dados, e garante que os resultados dos testes sejam confiáveis.
- Boas Práticas de Distribuição (PIB): Define os padrões de armazenamento, manuseio, e transporte de produtos farmacêuticos. O PIB garante que a qualidade dos medicamentos é mantida em toda a cadeia de abastecimento – por exemplo, controlando a temperatura, evitando contaminação, e rastreamento de lotes.
- Boas Práticas de Farmacovigilância (GVP): Fornece diretrizes para monitoramento da segurança de medicamentos e notificação de eventos adversos após a comercialização de um produto. O GVP ajuda empresas farmacêuticas e reguladores a detectar, avaliar, e prevenir efeitos colaterais negativos.
Cada área GxP é aplicada por regulamentos ou diretrizes específicas (Por exemplo, FDA 21 CFR para GMP/GLP/GCP, EU EudraLex for GMP/GDP/GVP, Eu diretrizes, QUEM, etc.). Embora os detalhes sejam diferentes, o objetivo comum é para garantir que os medicamentos sejam seguros, eficaz, e alta qualidade em cada etapa.
Mesa: Principais Disciplinas GxP(Tipos de regulamentos GxP)
| Tipo GxP |
Nome completo |
Estágio do ciclo de vida do produto |
Foco/Propósito |
| BPL |
Boas Práticas de Laboratório |
Pesquisa de drogas |
Governa estudos laboratoriais não clínicos (como testes de toxicidade) para garantir a qualidade e a rastreabilidade dos dados. GLP estabelece padrões para procedimentos de laboratório, gravação de dados, e relatórios. |
| GCP |
Boas Práticas de Laboratório |
Ensaios Clínicos |
Padrões internacionais para ensaios clínicos (I-GCP). Abrange o projeto de teste, consentimento informado, monitoramento, e relatórios precisos de resultados. |
| BPF |
Boas Práticas Clínicas |
Fabricação |
Garante que os medicamentos sejam produzidos e controlados de forma consistente na fabricação/embalagem. Inclui padrões de instalações/equipamentos, treinamento de pessoal, Teste de controle de qualidade. |
| PIB |
Boas Práticas de Distribuição |
Armazenagem & Logística |
Garante o armazenamento e transporte adequados de medicamentos (Por exemplo, temperatura correta, manuseio seguro) para que a qualidade seja mantida ao longo da cadeia de abastecimento. |
| GVP |
Boas Práticas de Farmacovigilância |
Monitoramento Pós-Mercado |
Diretrizes para monitoramento contínuo da segurança de medicamentos comercializados (notificação de eventos adversos, gestão de risco, comunicação com autoridades). |
Por que o GxP é importante na indústria farmacêutica?
A conformidade com GxP é essencial porque vidas literalmente dependem disso. Seus objetivos principais são:
- Segurança do Paciente: Seguir as GxP minimiza riscos como contaminação, confusões, ou erros de dosagem. Por exemplo, Regras de BPF (como saneamento adequado e controle de processo) ajudar a prevenir a contaminação cruzada entre produtos. Sem GxP, medicamentos de qualidade inferior podem prejudicar os pacientes ou não serem eficazes. Os órgãos reguladores enfatizam que “As regras GxP existem por um motivo: para proteger os pacientes”.
- Qualidade e consistência do produto: As estruturas GxP garantem que cada lote de um medicamento atenda às mesmas especificações. Isso inclui potência consistente, pureza, e estabilidade. Ao aplicar controles rigorosos (E.G.. calibração de instrumentos, métodos validados, testes em processo), os fabricantes podem garantir que cada frasco ou comprimido corresponde às reivindicações do rótulo.
- Conformidade Regulatória e Confiança: Aderir às GxP é legalmente obrigatório. Agências como FDA, Ema, MHRA, PMDA, e a OMS contam com inspeções e auditorias GxP. Empresas que demonstram fortes práticas de GxP constroem confiança com os reguladores. O não cumprimento pode levar a cartas de advertência, multas, recalls de produtos, ou pior. Como observa um guia da indústria, reguladores realizam auditorias para “verificar a conformidade com os padrões GxP”, o que ressalta a integridade do produto e a segurança do paciente.
- Integridade de dados: Um aspecto crítico do GxP é garantir que os dados sejam precisos e confiáveis. Regulamentações modernas (E.G.. FDA 21 Parte CFR 11, EU Annex 11) exigem que os registros eletrônicos sejam seguros, com data e hora, e à prova de adulteração. Princípios-chave como ALCOA+ (Atribuível, Legível, Contemporâneo, Original, Preciso, mais completo, Consistente, Duradouro, Disponível) são usados para julgar a qualidade dos dados. Na prática, isso significa implementar trilhas de auditoria em software, bloqueio de documentos mestre, e revisando regularmente os registros do lote. A integridade robusta dos dados é um pilar do GxP; sem isso, mesmo produtos bem feitos não seriam confiáveis.
Resumidamente, GxP é a base da gestão da qualidade farmacêutica. Ele garante que cada passo — desde os testes de laboratório até a embalagem final — é feito sob controle, condições documentadas. Por exemplo, um guia observa que “A conformidade com as GxP garante que medicamentos e produtos biológicos sejam desenvolvidos, fabricado, e testado de acordo com padrões rigorosos,” evitando contaminação dispendiosa ou erros de rotulagem. Por isso, As GxP não apenas protegem os pacientes, mas também sustentam uma reputação de marca confiável e acesso ao mercado para empresas farmacêuticas.

Compreendendo o GMP – O padrão GxP mais importante
Embora todas as áreas GxP sejam críticas, BPF (Boas Práticas de Fabricação) é frequentemente visto como a base para a conformidade da produção farmacêutica. GMP cobre todo o processo de fabricação de medicamentos, incluindo matérias-primas, equipamento, instalações, processos, e linhas de embalagem. Seu objetivo principal é minimizar o risco de contaminação ou desvio do produto, aplicando controles e documentação rigorosos. Os principais elementos do GMP incluem:
- Projeto de instalações e equipamentos: O equipamento deve ser projetado para operação higiênica. Isso significa usar materiais de qualidade farmacêutica (E.G.. 316L aço inoxidável), soldas lisas, sem zonas mortas, e fácil acesso para limpeza. Para máquinas de embalagem, isso pode implicar transportadores em balanço, peças de liberação rápida, e guardas fechados. O layout adequado evita confusões e facilita a validação da limpeza (procedimentos de limpeza validados).
- Utilitários e calibração: Todos os utilitários (água, ar comprimido, elétrico) deve atender às especificações de qualidade. Instrumentos e sensores (escalas, medidores de vazão, sondas de temperatura) requer regular calibração sob procedimentos escritos. A FDA espera explicitamente registros de calibração para equipamentos críticos. Por exemplo, uma linha de frasco cheio teria suas bombas volumétricas calibradas para garantir que cada dose fosse precisa.
- Validação e Qualificação: As GMP determinam que os equipamentos e processos sejam qualificados e validados. Isso envolve qualificação de instalação (QI), Qualificação Operacional (QO), e Qualificação de Desempenho (QP) para provar que o sistema funciona como pretendido (discutido em detalhes abaixo). Por exemplo, uma nova máquina de embalagem blister deve ser testada IQ/OQ/PQ para confirmar a temperatura de vedação, integridade do selo, e sistema de indexação atendem às especificações. Cada máquina crítica (preenchimentos de cápsula, cartonadores, linhas de enchimento de líquidos) deve seguir este ciclo de vida de validação.
- Procedimentos Operacionais Padrão (POPS): Detalhado, POPs escritos são obrigatórios para as etapas de fabricação, limpeza, manutenção, e controles de qualidade. Os operadores devem ser treinados nestes POPs e quaisquer alterações devem ser controladas através de um sistema formal de controle de alterações.. Os auditores procuram POPs e registros de treinamento atualizados.
- Boas Práticas de Documentação: GMP enfatiza que “se não estiver documentado, isso não aconteceu.” Todos os procedimentos, registros em lote, registros de limpeza, e os testes de CQ devem ser registrados de forma legível e contemporânea. Um registro de fabricação em lote concluído (TMB) deve incluir reconciliação de materiais, configurações do equipamento, verificações em processo, e quaisquer desvios/correções. Tendências modernas impulsionam registros eletrônicos de lote (eBR) com trilhas de auditoria sob 21 Parte CFR 11.
- Controle de qualidade (Controle de qualidade) e liberação de lote: Teste de controle de qualidade (E.G.. potência, esterilidade, identificação) é realizado em matérias-primas e produtos acabados. Somente após os departamentos de QA/QC revisarem toda a documentação e resultados de testes é que um lote será liberado. Esta verificação final é uma parte fundamental das BPF.
Blog da Jinlu Packing sobre embalagens GMP destaca vários desses requisitos. Por exemplo, observa que manutenção preventiva e calibração são exigidos pelo GMP (21 CFR 211.68 requer intervalos e registros definidos). Também enfatiza automação & prevenção de erros: modernas linhas de embalagem GMP usam inspeção visual, leitura de código de barras, e intertravamentos para evitar erros humanos. A integridade dos dados e os controles do computador também são cobertos pelas GMP: todos os sistemas informatizados na linha (E.G.. CLPs, sistemas de visão, HMIS) deve cumprir 21 Parte CFR 11 – significando logins exclusivos, trilhas de auditoria, assinaturas eletrônicas, e manutenção segura de registros. Na prática, isso significa que o software de controle da máquina registrará cada alteração de parâmetro com um carimbo de data/hora e ID do usuário, e exigirá a assinatura eletrônica de um gerente para aprovar um lote.
Lista de verificação de BPF para equipamentos de embalagem
Ajuda visualizar os requisitos de GMP em um formulário de lista de verificação. Para máquinas de embalagem, os fabricantes normalmente garantem:
- Projeto & Materiais: Construção higiênica (peças de aço inoxidável, Selos aprovados pela FDA), sem áreas “mortas”, superfícies inclinadas, fácil desmontagem para limpeza.
- Validação: Complete IQ/OQ/PQ em cada máquina. Teste documentado de funções (vedação, enchimento, pesagem) e desempenho. (Consulte a seção Qualificação do Equipamento abaixo.)
- Limpeza: Procedimentos de limpeza validados com testes de cotonete ou enxágue (normalmente usando TOC ou ensaios específicos) e registros.
- Manutenção & Calibração: Cronogramas de manutenção preventiva com registros. Calibração de bombas dosadoras, escalas, sensores para garantir a precisão.
- Controles & Automação: Sistemas de inspeção visual (Por exemplo, detectores de metais, controladores de peso) para detectar defeitos, sensores para evitar erros de alimentação, intertravamentos para parar se as portas abrirem.
- Integridade de dados: 21 Parte CFR 11 conformidade – trilhas de auditoria, controles de acesso do usuário, assinaturas eletrônicas, dados protegidos. Todos os registros (POPS, dados em lote, desvios) armazenado inalteravelmente.
- Rastreabilidade: Suporte para serialização/UIDs, código de barras, vinculando cada pacote primário a registros de lote, permitindo recall, se necessário. (As máquinas Jinlu geralmente integram impressoras de etiquetas ou leitores de código para rastreabilidade.)
- Ambiental & Controles de linha: Padrões apropriados para salas limpas, se necessário, verificações documentadas de liberação de linha entre lotes, rotulagem e manuseio de materiais adequados para evitar confusões.
Seguindo esses controles, os fabricantes podem ter certeza de que seus equipamentos de embalagem funcionarão dentro das diretrizes GMP. Por exemplo, Máquinas de preenchimento de cápsulas e Máquinas de embalagem de bolha da Jinlu são construídos com recursos prontos para GMP (superfícies lisas GMP, Capacidade CIP, etc.) para atender a essas demandas.

Requisitos GxP para fabricantes de equipamentos farmacêuticos
Os fornecedores de equipamentos de embalagem desempenham um papel vital no GxP. Os compradores esperam máquinas que não sejam apenas robustas e eficientes, mas também construído para facilitar a conformidade regulatória. Os principais requisitos incluem:
- Suporte de qualificação (IR/WH/PQ): Os fornecedores devem fornecer protocolos e assistência para qualificação. Isso significa qualificação de instalação documentada (QI) para mostrar que a máquina foi instalada corretamente, Qualificação Operacional (QO) para provar que funciona de acordo com as especificações, e Qualificação de Desempenho (QP) para verificar se produz consistentemente resultados aceitáveis. Por exemplo, A Jinlu Packing entrega cada máquina com um kit IQ/OQ/PQ e protocolos FAT/SAT completos. Muitas vezes, esses modelos podem ser adaptados pela equipe de controle de qualidade do comprador, economizando tempo durante a validação. O fornecedor também pode participar de execuções de qualificação ou fornecer listas de verificação de equipamentos certificados.
- Pacote de Documentação: Junto com a máquina, os fornecedores devem entregar um conjunto completo de documentos. Os itens típicos incluem Manual do usuário, Lista mestre de peças, Esquemas Elétricos, e Instruções de manutenção. Importante, GORDO (Teste de aceitação de fábrica) e SENTADO (Teste de aceitação do site) relatórios documentam que a máquina passou nos testes de fábrica e no local. Certificados de calibração para quaisquer dispositivos de medição devem ser incluídos. Na prática, um pacote de documentação compatível também listará as especificações do sistema, limpeza de POPs, avaliações de risco, e qualquer histórico de controle de alterações.
- Recursos de integridade de dados: Equipamentos modernos devem oferecer controles eletrônicos alinhados aos padrões de dados GxP. Isso inclui contas de usuário seguras (acesso baseado em função), assinaturas eletrônicas obrigatórias para ações críticas, e trilhas de auditoria completas de quaisquer alterações nos parâmetros. Por exemplo, uma tela HMI pode exigir o login do supervisor de turno para iniciar a produção, e cada alteração de receita ou configuração tem carimbo de data/hora. As máquinas também podem suportar saída digital de registros em lote, integração com sistemas MES/ERP.
- Ferramentas de validação e teste: Alguns fornecedores incluem ferramentas de software para registro de dados, calibração, ou validação. Este poderia ser um software pré-instalado para a realização de testes de sensores, ou funcionalidade integrada para bloquear parâmetros durante execuções PQ. Esses recursos reduzem o esforço manual durante a validação.
- Design Higiênico e de Segurança: Os equipamentos devem ser fáceis de limpar e manter. Recursos como peças de liberação rápida, zonas sem produto, e CIP (Limpeza no local) opções ajudam a atender às validações de limpeza. Os materiais em contato com o produto devem ser inertes (E.G.. 316LSS, Plásticos aprovados pela FDA). Protetores de segurança e intertravamentos protegem os operadores, mas também garantir o cumprimento (E.G.. máquina para quando aberta).
- Suporte pós-venda: A conformidade com GxP está em andamento. Os fabricantes podem exigir requalificação ou recalibração periódica. Os fornecedores devem oferecer serviços de ciclo de vida: peças de reposição (para substituição rápida de peças validadas), contratos de manutenção, e atualizações na documentação de validação se ocorrerem alterações. A disposição de um fornecedor em fornecer serviços de qualificação no local (IR/WH/PQ) pode facilitar muito os esforços de conformidade.
Lista de verificação de qualificação de equipamentos: A tabela abaixo resume etapas e documentos típicos para qualificar uma nova máquina farmacêutica:
| Fase |
Principais atividades |
Documentos Típicos |
| Requisitos do usuário (URS) |
Definir especificações críticas (E.G.. taxa de saída, precisão) |
Especificação de requisitos do usuário |
| Qualificação de Projeto (DQ) |
Verifique se o design do fornecedor atende ao URS |
Relatório de revisão de especificações de design |
| Aceitação de fábrica (GORDO) |
Testes de fábrica das principais funções, muitas vezes refletem testes de QI/QO |
Relatório FAT |
| Qualificação de Instalação (QI) |
Confirme a instalação correta: utilitários, configuração mecânica, documentação (desenhos, certificados) |
Protocolo de QI & lista de verificação |
| Qualificação Operacional (QO) |
Teste todas as funções: desempenho em vazio, controles, alarmes, recursos de segurança |
Protocolo OQ & resultados |
| Qualificação de Desempenho (QP) |
Execute a produção completa com produto real: verifique a qualidade da saída, consistência, condições de estresse |
Protocolo PQ, executar registros, resultados de testes de amostra |
| Lançamento Final |
Revise todos os registros de qualificação; Aprovação de controle de qualidade para colocar a máquina em produção GMP |
Relatório de resumo de qualificação |
(Observação: As máquinas de Jinlu vêm com Modelos de QI/OQ/PQ e um completo Aceitação de fábrica pacote de documentação, que os clientes podem adaptar conforme necessário.) Esta abordagem estruturada – desde os requisitos do usuário até a QP – é exigida por regulamentos como EU GMP Annex 15 e diretrizes da FDA. Pular qualquer etapa pode resultar em uma lacuna de conformidade.

Desafios comuns de conformidade com GxP
Mesmo com regras claras, as empresas muitas vezes enfrentam obstáculos na conformidade com GxP. Alguns desafios frequentes incluem:
- Validação ou documentação incompleta: Uma das lacunas mais graves é a utilização de equipamentos sem registros completos de IQ/OQ/PQ. “Pulando validação,”ou tendo parcial qualificação, é considerado um “lacuna fatal de BPF”. De forma similar, registros de lote e POPs ausentes ou desleixados prejudicam a conformidade: inspetores são treinados para procurar documentação. Como disse um especialista: “Se não estiver escrito, isso não aconteceu.” Má manutenção de registros (arquivos perdidos, notas ilegíveis, versões desatualizadas) é uma bandeira vermelha comum.
- Lapsos de manutenção e calibração de equipamentos: Os reguladores encontram repetidamente problemas como datas de vencimento de calibração expiradas ou reparos adiados. Um sensor quebrado ou uma balança não calibrada levará a dados ou produtos suspeitos. (Por exemplo, A análise de Sokol observa que calibrações esquecidas e peças desgastadas são “falhas simples” que pode desencadear uma retenção de lote.) Garantindo registros de manutenção rigorosos, usando rastreadores digitais para cronogramas de calibração, e capacitar a equipe para sinalizar problemas rapidamente são práticas recomendadas para superar isso.
- Problemas de integridade de dados: As linhas modernas de GMP dependem de sistemas eletrônicos. Falhas nos controles de dados podem quebrar o GxP. Os exemplos incluem trilhas de auditoria desabilitadas, senhas fracas, dados copiados/colados em vez de entradas originais, ou falha na revisão de registros eletrônicos. As empresas devem aplicar os princípios ALCOA+ — por exemplo,, certificando-se de que todas as entradas de dados sejam Atribuível (vinculado a um usuário), Legível (claro), Contemporâneo (gravado em tempo real), Original/Preciso, e Completo/Consistente. Treinar operadores e automatizar sempre que possível (E.G.. registros bloqueados por computador) ajudar a evitar substituições ou omissões manuais.
- Controle de mudanças e deficiências de CAPA: Um processo robusto de gerenciamento de mudanças é necessário para quaisquer mudanças (atualizações de equipamentos, novos POPs, novas matérias-primas). Um erro comum é não documentar uma alteração ou ignorar a revalidação após uma modificação. De forma similar, falha em investigar adequadamente os desvios (descartar os problemas como mero “erro humano” sem análise da causa raiz) pode deixar os problemas persistirem. Órgãos reguladores esperam forte CAPA (ação corretiva e preventiva) sistemas para resolver quaisquer desvios.
- Questões de Treinamento e Cultura: As GxP exigem que todo o pessoal seja treinado e esteja ciente dos procedimentos de qualidade. Programas de treinamento inadequados ou alta rotatividade podem levar a violações não intencionais. Construindo uma cultura de qualidade (onde a equipe se sente responsável pela conformidade e é incentivada a relatar problemas) é vital, mas muitas vezes lento para se desenvolver.
Resumindo, os desafios são muitas vezes organizacionais: documentação, manutenção, treinamento, e práticas de dados. Superá-los significa investir em sistemas (como software de gerenciamento eletrônico de documentos ou rastreamento de calibração), POPs disciplinados, e auditorias internas frequentes. As empresas que abordam proativamente essas áreas encontrarão a conformidade com as GxP mais tranquila durante as inspeções oficiais.
Como as empresas farmacêuticas alcançam a conformidade com GxP
Alcançar a conformidade com GxP é uma tarefa projeto abrangendo toda a organização. Abaixo está uma sequência típica de etapas (ilustrado no fluxograma) que uma empresa farmacêutica segue para construir um sistema compatível:
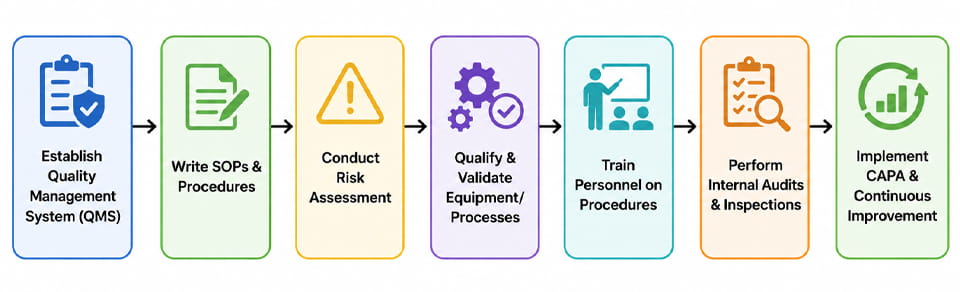
- Sistema de Gestão da Qualidade (SGQ): Comece definindo a estrutura organizacional para a qualidade (E.G.. manual de qualidade, políticas). Isso inclui atribuir funções e responsabilidades de qualidade.
- Desenvolva POPs & Documentação: Elaborar e aprovar procedimentos padrão para produção, teste, controle de mudanças, desvios, etc.. Certifique-se de que cada processo esteja claramente documentado.
- Avaliação de risco: Realize avaliações formais de risco (E.G.. FMEA) para identificar parâmetros e controles críticos do processo. Isso informa onde focar a validação e o monitoramento.
- Qualificação de Equipamento/Processo (IR/WH/PQ): Como na seção anterior, qualificar todos os equipamentos e processos de fabricação. Mantenha protocolos e relatórios de validação detalhados.
- Treinamento: Operadores de trem, Engenheiros, e equipe de QA/QC sobre os procedimentos aprovados, os princípios GxP, e o uso de equipamentos.
- Auditorias Internas: Realize autoinspeções de rotina ou auditorias simuladas para verificar a adesão e detectar problemas (E.G.. verificar registros de lote, registros ambientais, status de calibração).
- CAPA: Sempre que for identificado um desvio ou constatação (internamente ou por um regulador), investigar a causa raiz, aplicar ações corretivas, e atualizar procedimentos para evitar recorrência.
- Melhoria contínua: Usar dados (E.G.. análise de tendências de relatórios de fabricação ou registros de reclamações) para impulsionar melhorias de qualidade e otimizar processos.
Cada etapa retorna conforme necessário – por ex., uma mudança encontrada durante a auditoria leva à atualização dos POPs e à reciclagem. As empresas também usam o Gerenciamento de Risco de Qualidade (QRM) e Qualidade desde o Design (QbD) princípios para tornar esta abordagem sistemática. O fluxograma acima ilustra esse processo cíclico.
A relação entre GxP e equipamentos de embalagem
Moderno farmacêutico linhas de embalagem são complexos e devem incorporar os princípios GxP diretamente. Todas as máquinas da linha – desde descodificadoras de garrafas até máquinas de blister e encartuchadoras – devem ser projetadas e usadas de forma a respeitar as GMP. Aqui estão as principais conexões:
- Design Higiênico: As máquinas de embalagem são construídas para evitar contaminação. Por exemplo, um máquina de embalagem de bolha terá uma seção de formação fechada e trilhas de alimentação suaves, para que o produto não entre em contato com o chão ou superfícies empoeiradas. Ferramentas e peças para áreas de contato com produtos são normalmente de aço inoxidável ou plástico de uso médico, alinhamento com os requisitos de materiais GMP.
- Pronto para validação: O equipamento de embalagem deve ser totalmente qualificado. Os fornecedores geralmente projetam máquinas para fácil validação: áreas acessíveis para sensores (para verificações de calibração), capacidade de executar execuções vazias e completas, e desempenho estável. Os clientes esperam que as máquinas tenham especificações (POPs e manuais) que estão diretamente ligados às atividades de QI/OQ/PQ.
- Registros de lote e rastreabilidade: Cada etapa de uma linha GMP é documentada. As máquinas de embalagem automatizadas geralmente se integram ao software para registrar números de lote, velocidades de linha, e saída. Por exemplo, uma linha de engarrafamento pode rotular automaticamente cada garrafa com um código de lote e carimbo de data/hora. Esses códigos estão vinculados à execução de produção. Os sistemas também podem registrar peso ou contar rejeições (E.G.. cápsulas insuficientemente preenchidas sinalizadas por um controlador de peso em linha). Esses dados passam a fazer parte do registro eletrônico do lote. Resumidamente, máquinas de embalagem ajudam a manter rastreabilidade de cada unidade, que é um requisito regulatório.
- 21 Parte CFR 11 Conformidade: Como mencionado anteriormente, controles informatizados em equipamentos de embalagem (como a Interface Homem-Máquina e PLC) deve cumprir as regras de registro eletrônico. Isso significa que os operadores fazem login com IDs exclusivos, e nenhum parâmetro pode ser alterado sem autorização. Registros de dados (E.G.. configurações, resultados do teste) deve ser seguro e com data e hora. Muitas máquinas modernas agora incluem níveis de acesso de usuário (operador versus. supervisor) e recursos de registro de auditoria para atender a essas necessidades.
- Redução de erros e automação: A embalagem automatizada reduz os toques manuais, o que reduz o erro humano (um foco GxP). Por exemplo, Máquinas de preenchimento de cápsulas da Jinlu pode funcionar em alta velocidade com dosagem precisa, minimizando a necessidade de correção manual. Da mesma maneira, cartonadoras automatizadas garantir uma vedação consistente. Para conformidade, isso significa menos chances de componentes mal escolhidos ou rótulos errados – crucial ao embalar diferentes produtos lado a lado.
- Recursos regulatórios: Novos regulamentos como DSCSA (EUA) ou febre aftosa (UE) exigir números de série em embalagens individuais. As máquinas de embalagem são frequentemente equipadas com impressoras de códigos 2D e câmeras de visão para aplicar/verificar esses códigos. Tais características mostram como as leis do mercado (em nome da rastreabilidade GxP) funções do equipamento de forma.
- Suporte de conformidade: Equipamento de Jinlu, por exemplo, está pronto para GMP e geralmente vem com suporte para qualificação e rastreabilidade. Um enchimento de cápsula típico pode apresentar um CIP (limpar no local) sistema e um funil de alimentação removível para esterilização. UM linha de bolha pode incluir portas de proteção com travas de segurança para impedir a operação quando abertas. Esses detalhes de design suportam diretamente o GMP.
Ao escolher máquinas de embalagem com GxP em mente, empresas farmacêuticas tornam a conformidade mais fácil. Por exemplo, instalando um Jinlu máquina de enchimento de cápsulas ou máquina de encadernação significa que o comprador já possui uma máquina construída de acordo com os padrões farmacêuticos, com documentação (como FAT/SAT) pronto para validação. Em última análise, equipamentos bem projetados são a base de um processo de fabricação compatível.

Conclusão
GxP é a base da qualidade farmacêutica. Não é apenas um conjunto de regras, mas um compromisso de toda a empresa para tornar seguro, medicamentos eficazes. Em sua essência, GxP garante que “os produtos farmacêuticos são corrigidos.” Boas Práticas de Fabricação (BPF) é a parte mais proeminente do GxP para produção de medicamentos, cobrindo design de equipamentos higiênicos, processos validados, e documentação rigorosa. Outros componentes como GLP, GCP, PIB, e GVP abordam diferentes estágios (estudos de laboratório, testes, distribuição, e farmacovigilância, respectivamente), mas todos partilham o objectivo de proteger os pacientes.
Equipamento de embalagem farmacêutica desempenha um papel vital na conformidade com GxP. Máquinas como enchedoras de cápsulas, embaladores de bolhas, e as encartuchadeiras devem ser construídas e validadas para atender aos padrões GMP – por exemplo, sendo fácil de limpar, suporte a registros eletrônicos de lote, e mantendo a rastreabilidade. Ao escolher Maquinário pronto para GMP e seguindo protocolos de qualificação (IR/WH/PQ), as empresas podem integrar os princípios GxP em suas linhas de produção.
Pronto para garantir conformidade com GxP em sua linha de produção? Considere o equipamento de embalagem avançado da Jinlu Packing, que é projetado para ambientes GMP. Contato Jinlu para solicitar um orçamento ou saber mais sobre nossas máquinas de envase de cápsulas em conformidade com GMP, embaladores de bolhas, e linhas de embalagem.
Perguntas frequentes sobre GxP na indústria farmacêutica
O que significa GxP?
GxP significa “Boas Práticas”. É um termo geral para várias diretrizes de qualidade em produtos farmacêuticos.. O “x” pode ser M (Fabricação), eu (Laboratório), C (Clínico), D (Distribuição), etc.. Em outras palavras, Boas Práticas de Fabricação (BPF), Boas Práticas de Laboratório (BPL), Boas Práticas Clínicas (GCP), etc..
O GMP faz parte do GxP?
Sim. BPF (Boas Práticas de Fabricação) é um dos principais componentes do GxP. GxP é o guarda-chuva, e GMP refere-se especificamente à fabricação. Então, quando falamos sobre conformidade com GxP, As BPF são muitas vezes o maior foco porque regem a produção e a embalagem. Outras partes como GLP ou GVP também estão na família GxP.
Qual é a diferença entre GxP e GMP?
GxP é a categoria ampla de todas as “Boas Práticas” no setor farmacêutico (cobrindo o desenvolvimento através da distribuição). GMP é apenas uma dessas práticas, focado na produção. Pense no GxP como toda a estrutura de qualidade, e GMP como a seção que trata dos padrões de chão de fábrica.
Quem regula a conformidade com GxP?
Agências reguladoras aplicam GxP. Nos EUA, o FDA supervisiona cGMP e GLP, e FDA/NIH supervisionam GCP em ensaios clínicos. Na Europa, Ema (e órgãos nacionais como MHRA) aplicar as diretrizes GMP e GCP da UE. A OMS publica orientações internacionais sobre GxP que muitos países adotam. Cada país pode ter sua própria versão, mas FDA, Ema, A OMS é a principal autoridade referenciada globalmente.
O que é 21 Parte CFR 11 e por que isso é importante para o GxP?
21 Parte CFR 11 é uma regulamentação da FDA dos EUA sobre registros eletrônicos e assinaturas eletrônicas. Para GxP, significa qualquer sistema informatizado que gere registros (como uma IHM de máquina ou software LIMS) deve ter controles para que os registros sejam seguros, com data e hora, e não pode ser adulterado. Por exemplo, o sistema de controle de uma máquina de embalagem blister precisará de logins seguros e uma trilha de auditoria para alterações de parâmetros. Conformidade com a Parte 11 é obrigatório nos EUA e orienta outras regiões (EU Annex 11) em sistemas eletrônicos.
As máquinas de embalagem farmacêutica precisam estar em conformidade com as GxP?
Absolutamente. Qualquer equipamento usado para fabricar ou embalar medicamentos deve atender aos critérios de projeto e validação GxP. Isto significa máquinas de embalagem (preenchimentos de cápsula, máquinas de bolha, enchimentos líquidos, cartonadores, etc.) deveria ter design higiênico, operação validada, e recursos de integridade de dados. Por exemplo, uma linha de embalagem deve seguir procedimentos de limpeza documentados (um requisito de BPF) e usar sistemas que registram dados em lote. Em muitos casos, fornecedores de equipamentos de embalagem anunciam máquinas “compatíveis com GMP” ou “prontas para FDA”. Ao selecionar o equipamento, os fabricantes pedem recursos como peças de contato em aço inoxidável, limpeza fácil, e protocolos IQ/OQ/PQ completos. Equipamento de Jinlu, por exemplo, foi projetado com esses padrões em mente para oferecer suporte a um ambiente regulamentado por GxP.
Referências:
1.Boas Práticas de Fabricação -- QUEM
2.TRS 986 – Anexo 2: Boas práticas de fabricação da OMS para produtos farmacêuticos: Princípios principais -- QUEM
3.Diretrizes da OMS para Produção Farmacêutica -- QUEM
4.Orientação sobre boas práticas de fabricação e boas práticas de distribuição: Perguntas e respostas —— Agência Europeia de Medicamentos
5.Orientação MHRA sobre integridade de dados GxP —— gov.uk
6.Guia de boas práticas do GAMP: Operação de Sistemas Informatizados GxP —— ispe.org




